Forced vital capacity (FVC) Best measure - all weights
wesleycrouse
2022-02-28
Last updated: 2022-03-03
Checks: 6 1
Knit directory: ctwas_applied/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210726) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 380982d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Unstaged changes:
Modified: analysis/ukb-a-232_allweights.Rmd
Modified: analysis/ukb-a-249_allweights.Rmd
Modified: analysis/ukb-a-389_allweights.Rmd
Modified: analysis/ukb-a-66_allweights.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ukb-a-232_allweights.Rmd) and HTML (docs/ukb-a-232_allweights.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 380982d | wesleycrouse | 2022-03-01 | fixing typo in all weight reports |
| Rmd | 76fa2cd | wesleycrouse | 2022-03-01 | cleaning up all weight reports |
| html | 76fa2cd | wesleycrouse | 2022-03-01 | cleaning up all weight reports |
| html | 2509c32 | wesleycrouse | 2022-03-01 | additional traits for all weight analysis |
| Rmd | 962fd16 | wesleycrouse | 2022-03-01 | additional traits for all weight analysis |
trait_id <- "ukb-a-232"
trait_name <- "Forced vital capacity (FVC) Best measure"
source("/project2/mstephens/wcrouse/UKB_analysis_allweights/ctwas_config.R")
trait_dir <- paste0("/project2/mstephens/wcrouse/UKB_analysis_allweights/", trait_id)
results_dirs <- list.dirs(trait_dir, recursive=F)Load cTWAS results for all weights
# df <- list()
#
# for (i in 1:length(results_dirs)){
# #print(i)
#
# results_dir <- results_dirs[i]
# weight <- rev(unlist(strsplit(results_dir, "/")))[1]
# analysis_id <- paste(trait_id, weight, sep="_")
#
# #load ctwas results
# ctwas_res <- data.table::fread(paste0(results_dir, "/", analysis_id, "_ctwas.susieIrss.txt"))
#
# #load z scores for SNPs and collect sample size
# load(paste0(results_dir, "/", analysis_id, "_expr_z_snp.Rd"))
#
# sample_size <- z_snp$ss
# sample_size <- as.numeric(names(which.max(table(sample_size))))
#
# #separate gene and SNP results
# ctwas_gene_res <- ctwas_res[ctwas_res$type == "gene", ]
# ctwas_gene_res <- data.frame(ctwas_gene_res)
# ctwas_snp_res <- ctwas_res[ctwas_res$type == "SNP", ]
# ctwas_snp_res <- data.frame(ctwas_snp_res)
#
# #add gene information to results
# sqlite <- RSQLite::dbDriver("SQLite")
# db = RSQLite::dbConnect(sqlite, paste0("/project2/compbio/predictdb/mashr_models/mashr_", weight, ".db"))
# query <- function(...) RSQLite::dbGetQuery(db, ...)
# gene_info <- query("select gene, genename, gene_type from extra")
# RSQLite::dbDisconnect(db)
#
# ctwas_gene_res <- cbind(ctwas_gene_res, gene_info[sapply(ctwas_gene_res$id, match, gene_info$gene), c("genename", "gene_type")])
#
# #add z scores to results
# load(paste0(results_dir, "/", analysis_id, "_expr_z_gene.Rd"))
# ctwas_gene_res$z <- z_gene[ctwas_gene_res$id,]$z
#
# z_snp <- z_snp[z_snp$id %in% ctwas_snp_res$id,]
# ctwas_snp_res$z <- z_snp$z[match(ctwas_snp_res$id, z_snp$id)]
#
# #merge gene and snp results with added information
# ctwas_snp_res$genename=NA
# ctwas_snp_res$gene_type=NA
#
# ctwas_res <- rbind(ctwas_gene_res,
# ctwas_snp_res[,colnames(ctwas_gene_res)])
#
# #get number of SNPs from s1 results; adjust for thin argument
# ctwas_res_s1 <- data.table::fread(paste0(results_dir, "/", analysis_id, "_ctwas.s1.susieIrss.txt"))
# n_snps <- sum(ctwas_res_s1$type=="SNP")/thin
# rm(ctwas_res_s1)
#
# #load estimated parameters
# load(paste0(results_dir, "/", analysis_id, "_ctwas.s2.susieIrssres.Rd"))
#
# #estimated group prior
# estimated_group_prior <- group_prior_rec[,ncol(group_prior_rec)]
# names(estimated_group_prior) <- c("gene", "snp")
# estimated_group_prior["snp"] <- estimated_group_prior["snp"]*thin #adjust parameter to account for thin argument
#
# #estimated group prior variance
# estimated_group_prior_var <- group_prior_var_rec[,ncol(group_prior_var_rec)]
# names(estimated_group_prior_var) <- c("gene", "snp")
#
# #report group size
# group_size <- c(nrow(ctwas_gene_res), n_snps)
#
# #estimated group PVE
# estimated_group_pve <- estimated_group_prior_var*estimated_group_prior*group_size/sample_size
# names(estimated_group_pve) <- c("gene", "snp")
#
# #ctwas genes using PIP>0.8
# ctwas_genes <- ctwas_gene_res$genename[ctwas_gene_res$susie_pip>0.8]
#
# #twas genes using bonferroni threshold
# alpha <- 0.05
# sig_thresh <- qnorm(1-(alpha/nrow(ctwas_gene_res)/2), lower=T)
# twas_genes <- ctwas_gene_res$genename[abs(ctwas_gene_res$z) > sig_thresh]
#
#
# df[[weight]] <- list(prior=estimated_group_prior,
# prior_var=estimated_group_prior_var,
# pve=estimated_group_pve,
# ctwas=ctwas_genes,
# twas=twas_genes )
# }
#
# save(df, file=paste(trait_dir, "results_df.RData", sep="/"))
load(paste(trait_dir, "results_df.RData", sep="/"))
output <- data.frame(weight=names(df),
prior_g=unlist(lapply(df, function(x){x$prior["gene"]})),
prior_s=unlist(lapply(df, function(x){x$prior["snp"]})),
prior_var_g=unlist(lapply(df, function(x){x$prior_var["gene"]})),
prior_var_s=unlist(lapply(df, function(x){x$prior_var["snp"]})),
pve_g=unlist(lapply(df, function(x){x$pve["gene"]})),
pve_s=unlist(lapply(df, function(x){x$pve["snp"]})),
n_ctwas=unlist(lapply(df, function(x){length(x$ctwas)})),
n_twas=unlist(lapply(df, function(x){length(x$twas)})),
row.names=NULL)Plot estimated prior parameters and PVE
#plot estimated group prior
output <- output[order(-output$prior_g),]
par(mar=c(10.1, 4.1, 4.1, 2.1))
plot(output$prior_g, type="l", ylim=c(0, max(output$prior_g, output$prior_s)*1.1),
xlab="", ylab="Estimated Group Prior", xaxt = "n", col="blue")
lines(output$prior_s)
axis(1, at = 1:nrow(output),
labels = output$weight,
las=2,
cex.axis=0.6)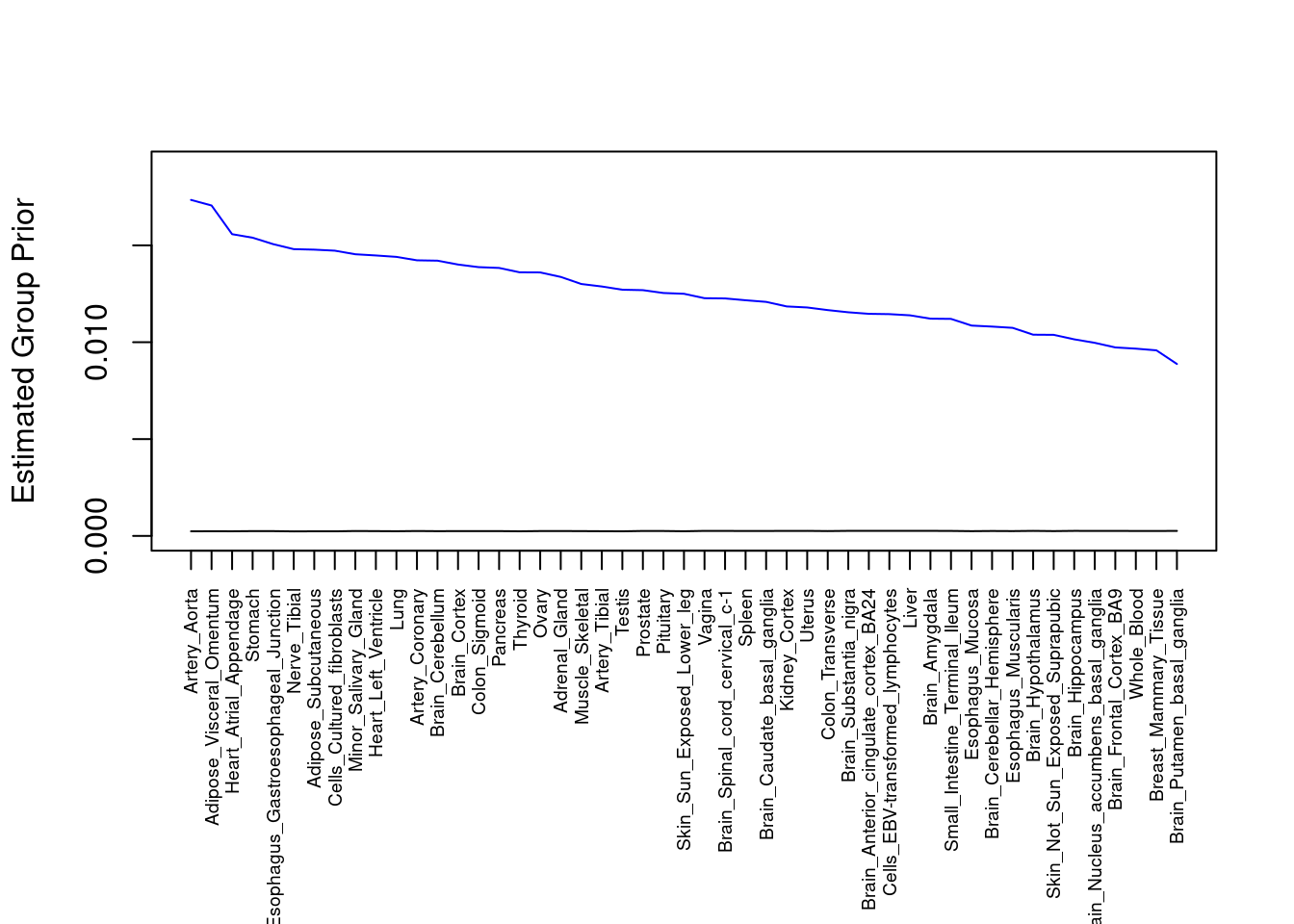
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
####################
#plot estimated group prior variance
par(mar=c(10.1, 4.1, 4.1, 2.1))
plot(output$prior_var_g, type="l", ylim=c(0, max(output$prior_var_g, output$prior_var_s)*1.1),
xlab="", ylab="Estimated Group Prior Variance", xaxt = "n", col="blue")
lines(output$prior_var_s)
axis(1, at = 1:nrow(output),
labels = output$weight,
las=2,
cex.axis=0.6)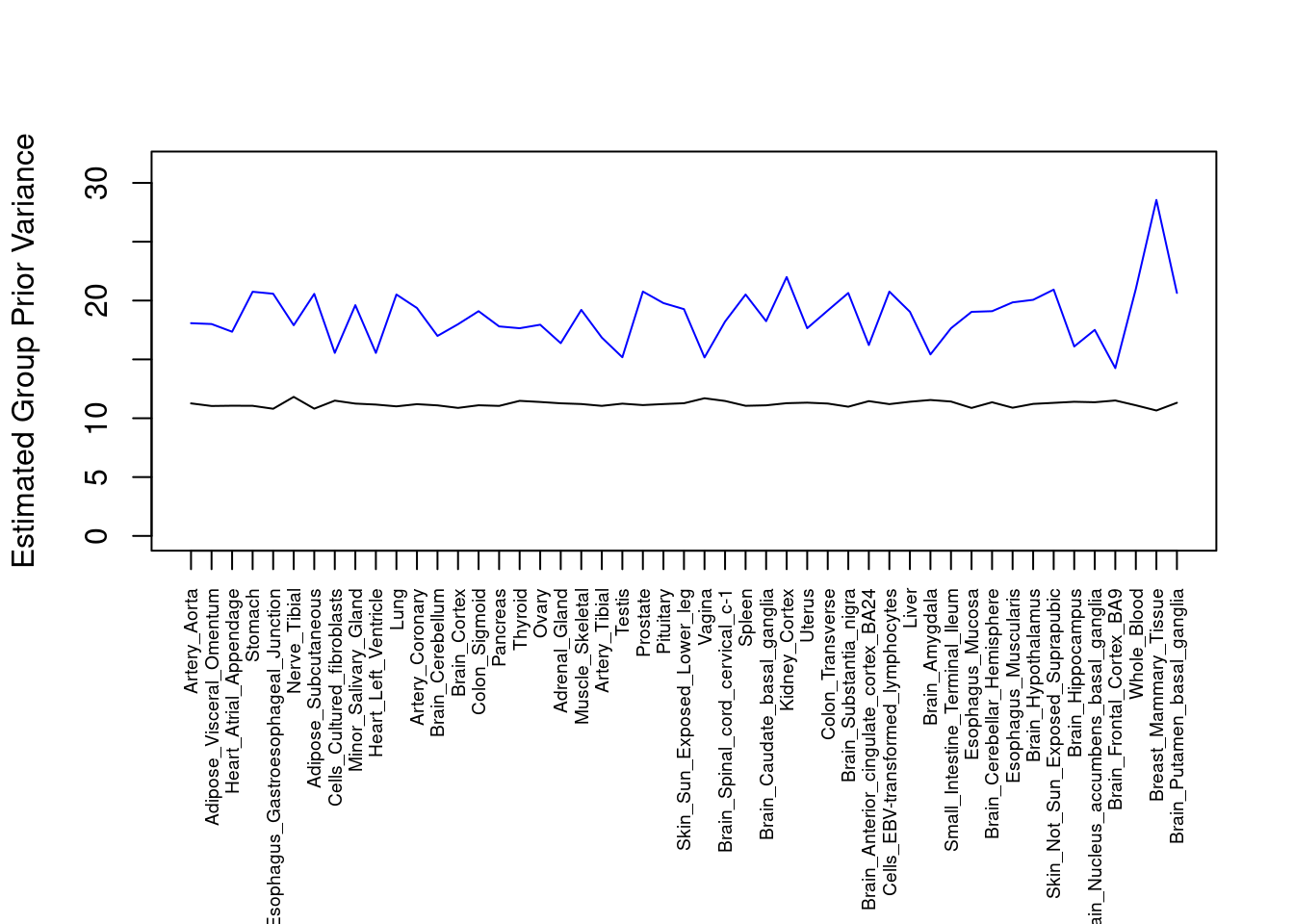
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
####################
#plot PVE
output <- output[order(-output$pve_g),]
par(mar=c(10.1, 4.1, 4.1, 2.1))
#plot(output$pve_g, type="l", ylim=c(0, max(output$pve_g, output$pve_s)*1.1),
plot(output$pve_g, type="l", ylim=c(0, max(output$pve_g+output$pve_s)*1.1),
xlab="", ylab="Estimated PVE", xaxt = "n", col="blue")
lines(output$pve_s)
lines(output$pve_g+output$pve_s, lty=2)
axis(1, at = 1:nrow(output),
labels = output$weight,
las=2,
cex.axis=0.6)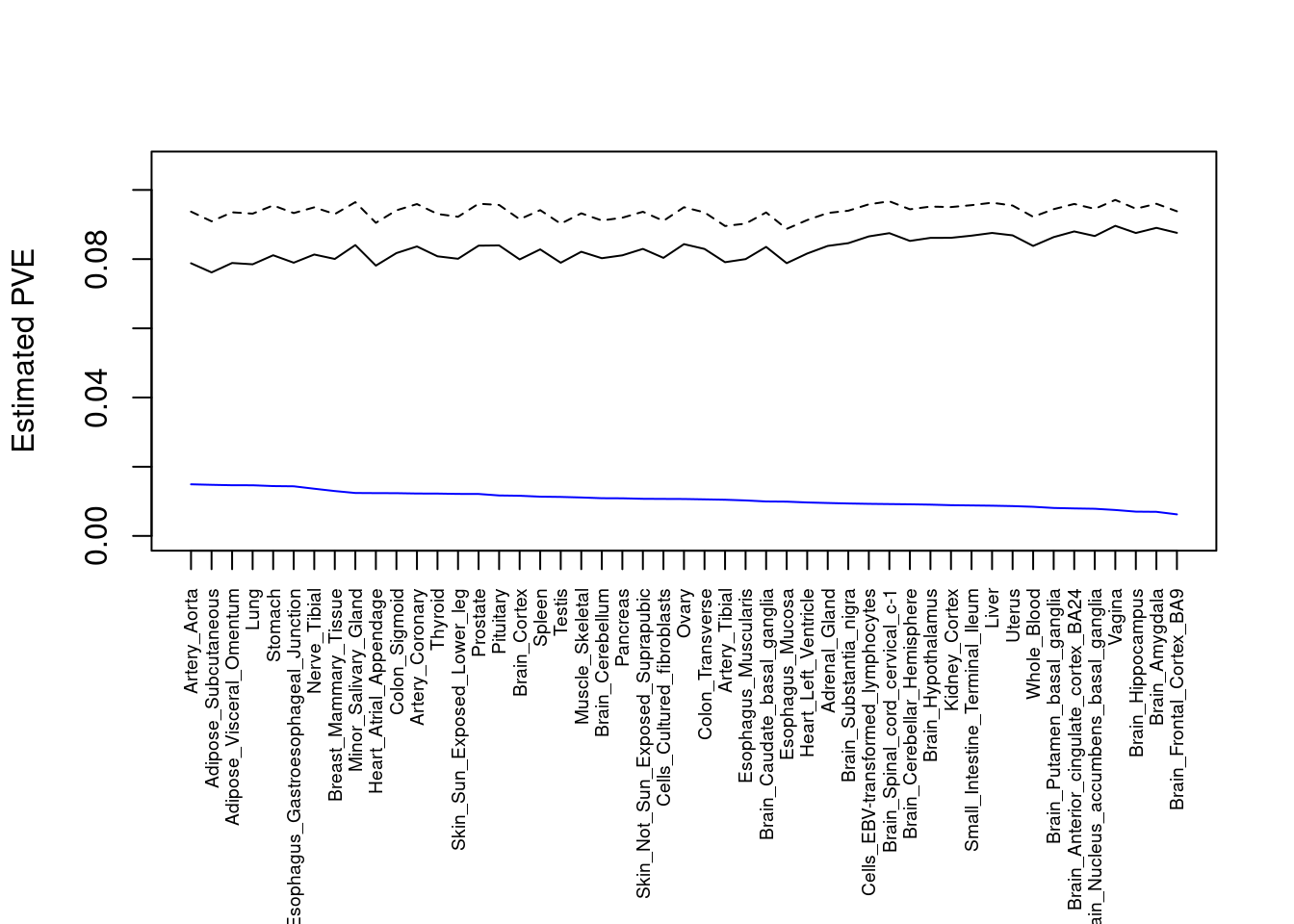
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
Number of cTWAS and TWAS genes
cTWAS genes are the set of genes with PIP>0.8 in any tissue. TWAS genes are the set of genes with significant z score (Bonferroni within tissue) in any tissue.
#plot number of significant cTWAS and TWAS genes in each tissue
plot(output$n_ctwas, output$n_twas, xlab="Number of cTWAS Genes", ylab="Number of TWAS Genes")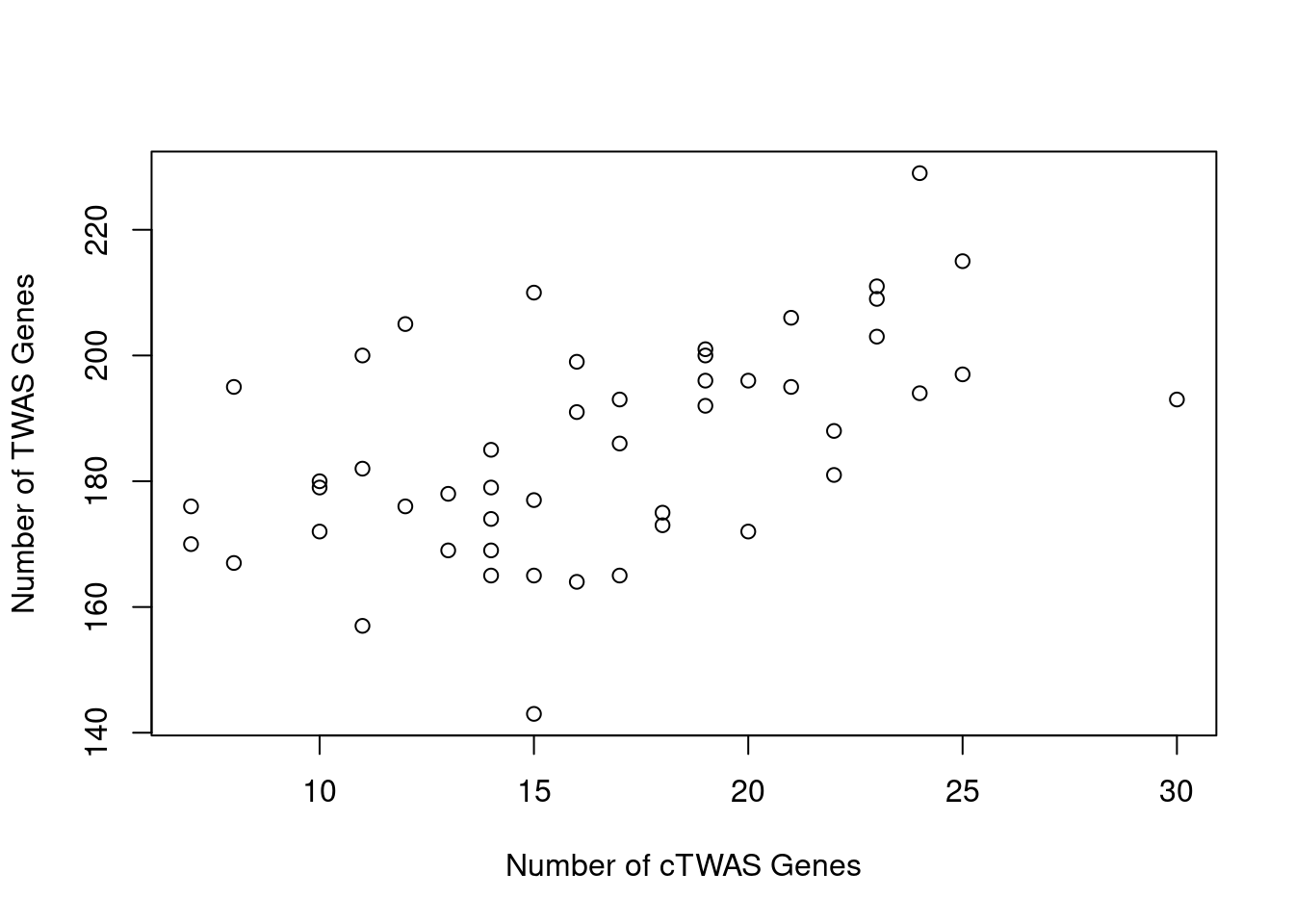
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
#number of ctwas_genes
ctwas_genes <- unique(unlist(lapply(df, function(x){x$ctwas})))
length(ctwas_genes)[1] 227#number of twas_genes
twas_genes <- unique(unlist(lapply(df, function(x){x$twas})))
length(twas_genes)[1] 995Enrichment analysis for cTWAS genes
#enrichment for cTWAS genes
library(enrichR)Welcome to enrichR
Checking connection ... Enrichr ... Connection is Live!
FlyEnrichr ... Connection is available!
WormEnrichr ... Connection is available!
YeastEnrichr ... Connection is available!
FishEnrichr ... Connection is available!dbs <- c("GO_Biological_Process_2021", "GO_Cellular_Component_2021", "GO_Molecular_Function_2021")
GO_enrichment <- enrichr(ctwas_genes, dbs)Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.for (db in dbs){
print(db)
enrich_results <- GO_enrichment[[db]]
print(plotEnrich(GO_enrichment[[db]]))
enrich_results <- enrich_results[enrich_results$Adjusted.P.value<0.05,c("Term", "Overlap", "Adjusted.P.value", "Genes")]
print(enrich_results)
}[1] "GO_Biological_Process_2021"
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)Enrichment analysis for TWAS genes
#enrichment for TWAS genes
dbs <- c("GO_Biological_Process_2021", "GO_Cellular_Component_2021", "GO_Molecular_Function_2021")
GO_enrichment <- enrichr(twas_genes, dbs)Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.for (db in dbs){
print(db)
enrich_results <- GO_enrichment[[db]]
print(plotEnrich(GO_enrichment[[db]]))
enrich_results <- enrich_results[enrich_results$Adjusted.P.value<0.05,c("Term", "Overlap", "Adjusted.P.value", "Genes")]
print(enrich_results)
}[1] "GO_Biological_Process_2021"
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"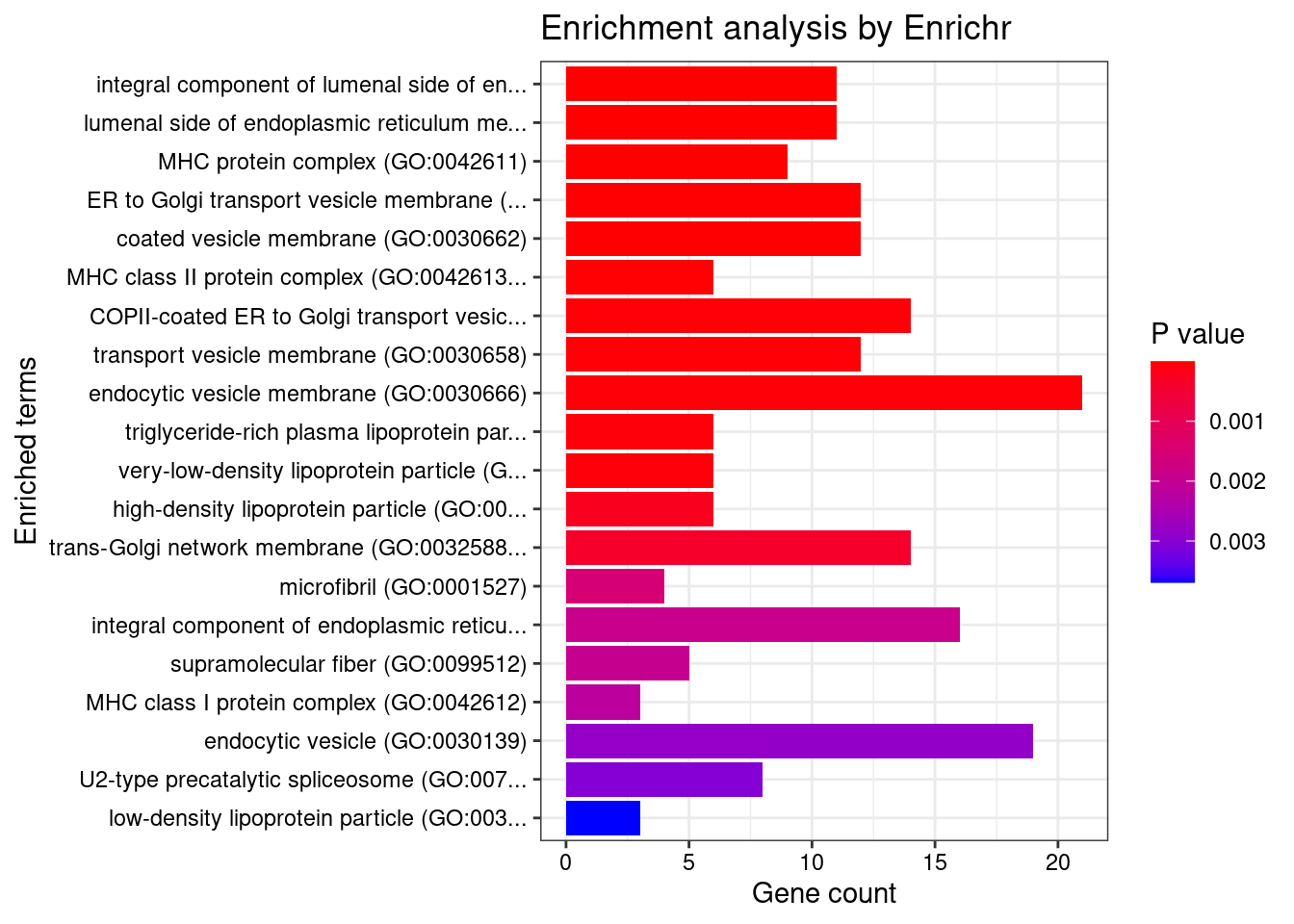
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
Term
1 integral component of lumenal side of endoplasmic reticulum membrane (GO:0071556)
2 lumenal side of endoplasmic reticulum membrane (GO:0098553)
3 MHC protein complex (GO:0042611)
4 ER to Golgi transport vesicle membrane (GO:0012507)
5 coated vesicle membrane (GO:0030662)
6 MHC class II protein complex (GO:0042613)
7 COPII-coated ER to Golgi transport vesicle (GO:0030134)
8 transport vesicle membrane (GO:0030658)
9 endocytic vesicle membrane (GO:0030666)
10 triglyceride-rich plasma lipoprotein particle (GO:0034385)
11 very-low-density lipoprotein particle (GO:0034361)
12 high-density lipoprotein particle (GO:0034364)
13 trans-Golgi network membrane (GO:0032588)
14 microfibril (GO:0001527)
15 integral component of endoplasmic reticulum membrane (GO:0030176)
16 supramolecular fiber (GO:0099512)
17 MHC class I protein complex (GO:0042612)
18 endocytic vesicle (GO:0030139)
19 U2-type precatalytic spliceosome (GO:0071005)
Overlap Adjusted.P.value
1 11/28 6.580973e-06
2 11/28 6.580973e-06
3 9/20 1.877092e-05
4 12/54 7.947428e-04
5 12/55 7.947428e-04
6 6/13 9.665871e-04
7 14/79 1.259659e-03
8 12/60 1.259659e-03
9 21/158 1.305060e-03
10 6/15 1.410980e-03
11 6/15 1.410980e-03
12 6/19 5.906028e-03
13 14/99 8.972080e-03
14 4/11 3.314618e-02
15 16/142 3.739480e-02
16 5/19 3.739480e-02
17 3/6 3.945998e-02
18 19/189 4.851940e-02
19 8/50 4.910404e-02
Genes
1 HLA-DRB5;SPPL2C;HLA-B;HLA-C;HLA-DRA;HLA-DQA2;HLA-G;HLA-DQA1;HLA-DQB2;HLA-DRB1;HLA-DQB1
2 HLA-DRB5;SPPL2C;HLA-B;HLA-C;HLA-DRA;HLA-DQA2;HLA-G;HLA-DQA1;HLA-DQB2;HLA-DRB1;HLA-DQB1
3 HLA-DRB5;HFE;HLA-B;HLA-C;HLA-DRA;HLA-DQA1;HLA-DQB2;HLA-DRB1;HLA-DQB1
4 HLA-DRB5;SAR1B;HLA-B;HLA-C;HLA-DRA;HLA-G;HLA-DQA2;SEC24C;HLA-DQB2;HLA-DQA1;HLA-DRB1;HLA-DQB1
5 HLA-DRB5;SAR1B;HLA-B;HLA-C;HLA-DRA;HLA-G;HLA-DQA2;SEC24C;HLA-DQB2;HLA-DQA1;HLA-DRB1;HLA-DQB1
6 HLA-DRB5;HLA-DRA;HLA-DQA1;HLA-DQB2;HLA-DRB1;HLA-DQB1
7 HLA-DRB5;SAR1B;HLA-B;HLA-C;HLA-G;HLA-DRA;TMED6;ERGIC2;HLA-DQA2;SEC24C;HLA-DQB2;HLA-DQA1;HLA-DRB1;HLA-DQB1
8 HLA-DRB5;SAR1B;HLA-B;HLA-C;HLA-DRA;HLA-G;HLA-DQA2;SEC24C;HLA-DQB2;HLA-DQA1;HLA-DRB1;HLA-DQB1
9 CAMK2B;HLA-DRB5;PTCH1;HLA-B;TAP2;HLA-C;TAP1;TCIRG1;HLA-G;DNM2;NOSTRIN;HLA-DRA;STX4;APOE;HLA-DQA2;HLA-DQA1;WNT3;HLA-DRB1;HLA-DQB2;WNT4;HLA-DQB1
10 APOM;APOC1;APOC4;APOE;APOA5;PCYOX1
11 APOM;APOC1;APOC4;APOE;APOA5;PCYOX1
12 APOM;APOC1;APOC4;APOE;APOA5;PLTP
13 COG8;HLA-DRB5;COG4;AP4E1;BAIAP3;ARFRP1;AP1S1;HLA-DRA;HLA-DQA2;HLA-DQA1;HLA-DRB1;HLA-DQB2;BOK;HLA-DQB1
14 FBN2;MFAP2;LTBP4;ADAMTS10
15 HLA-DRB5;CAMLG;ATF6B;SPPL2C;CCDC47;HLA-B;TAP2;HLA-C;TAP1;HLA-G;HLA-DRA;HLA-DQA2;HLA-DQA1;HLA-DRB1;HLA-DQB2;HLA-DQB1
16 FBN2;ELN;MFAP2;LTBP4;ADAMTS10
17 HFE;HLA-B;HLA-C
18 CAMK2B;RAB5B;HLA-DRB5;PTCH1;LPAR1;DYSF;DNM2;RAB24;NOSTRIN;HLA-DRA;RIN3;APOE;HLA-DQA2;HLA-DQA1;WNT3;HLA-DRB1;HLA-DQB2;WNT4;HLA-DQB1
19 EFTUD2;SF3A3;SART1;IK;SF3B2;SNRPD2;ZMAT2;SNRPG
[1] "GO_Molecular_Function_2021"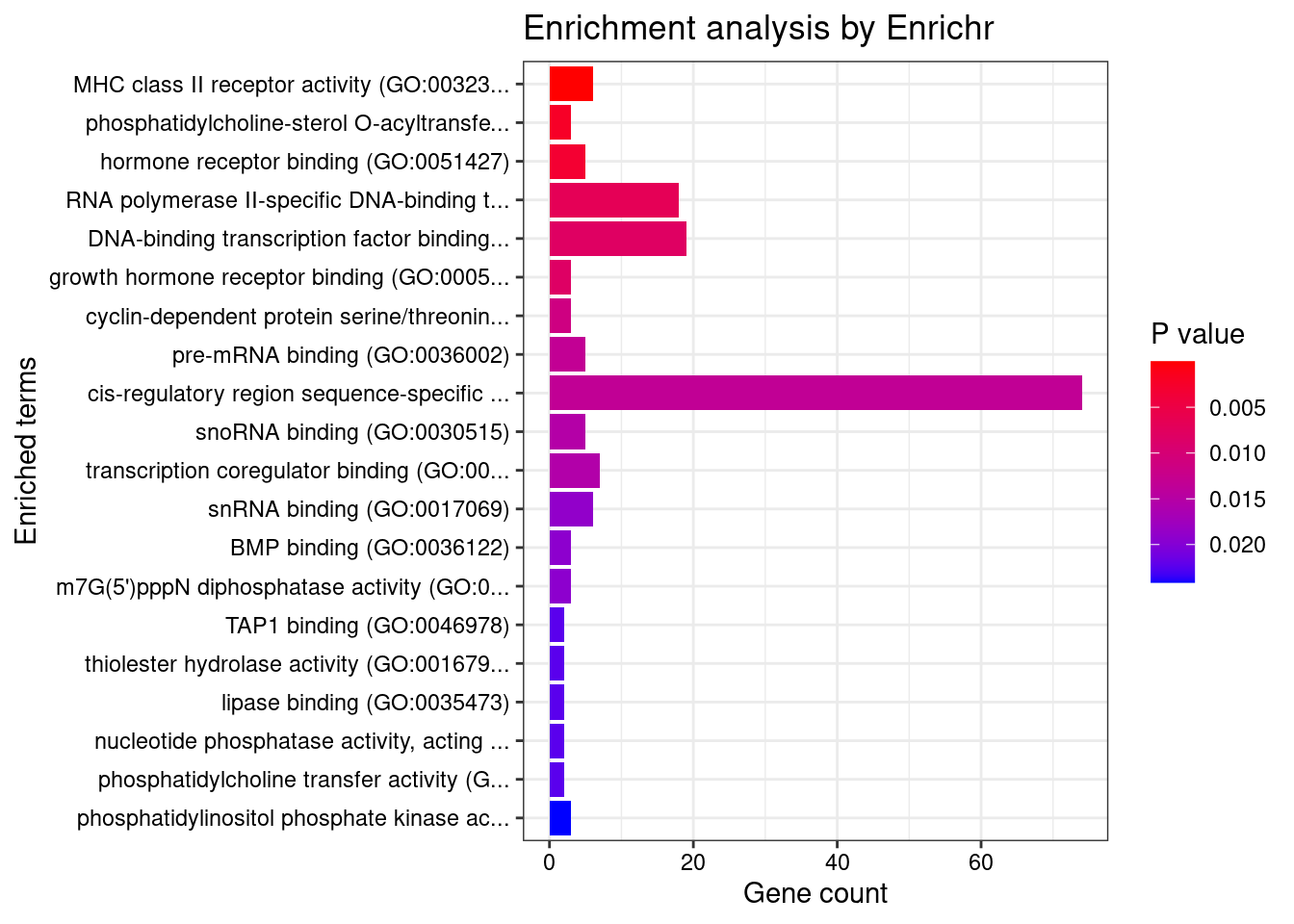
| Version | Author | Date |
|---|---|---|
| 2509c32 | wesleycrouse | 2022-03-01 |
Term Overlap Adjusted.P.value
1 MHC class II receptor activity (GO:0032395) 6/10 0.001583806
Genes
1 HLA-DRA;HLA-DQA2;HLA-DQA1;HLA-DQB2;HLA-DRB1;HLA-DQB1Enrichment analysis for cTWAS genes in top tissues separately
output <- output[order(-output$pve_g),]
top_tissues <- output$weight[1:5]
for (tissue in top_tissues){
ctwas_genes_tissue <- df[[tissue]]$ctwas
dbs <- c("GO_Biological_Process_2021", "GO_Cellular_Component_2021", "GO_Molecular_Function_2021")
GO_enrichment <- enrichr(ctwas_genes_tissue, dbs)
for (db in dbs){
print(db)
enrich_results <- GO_enrichment[[db]]
print(plotEnrich(GO_enrichment[[db]]))
enrich_results <- enrich_results[enrich_results$Adjusted.P.value<0.05,c("Term", "Overlap", "Adjusted.P.value", "Genes")]
print(enrich_results)
}
}Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"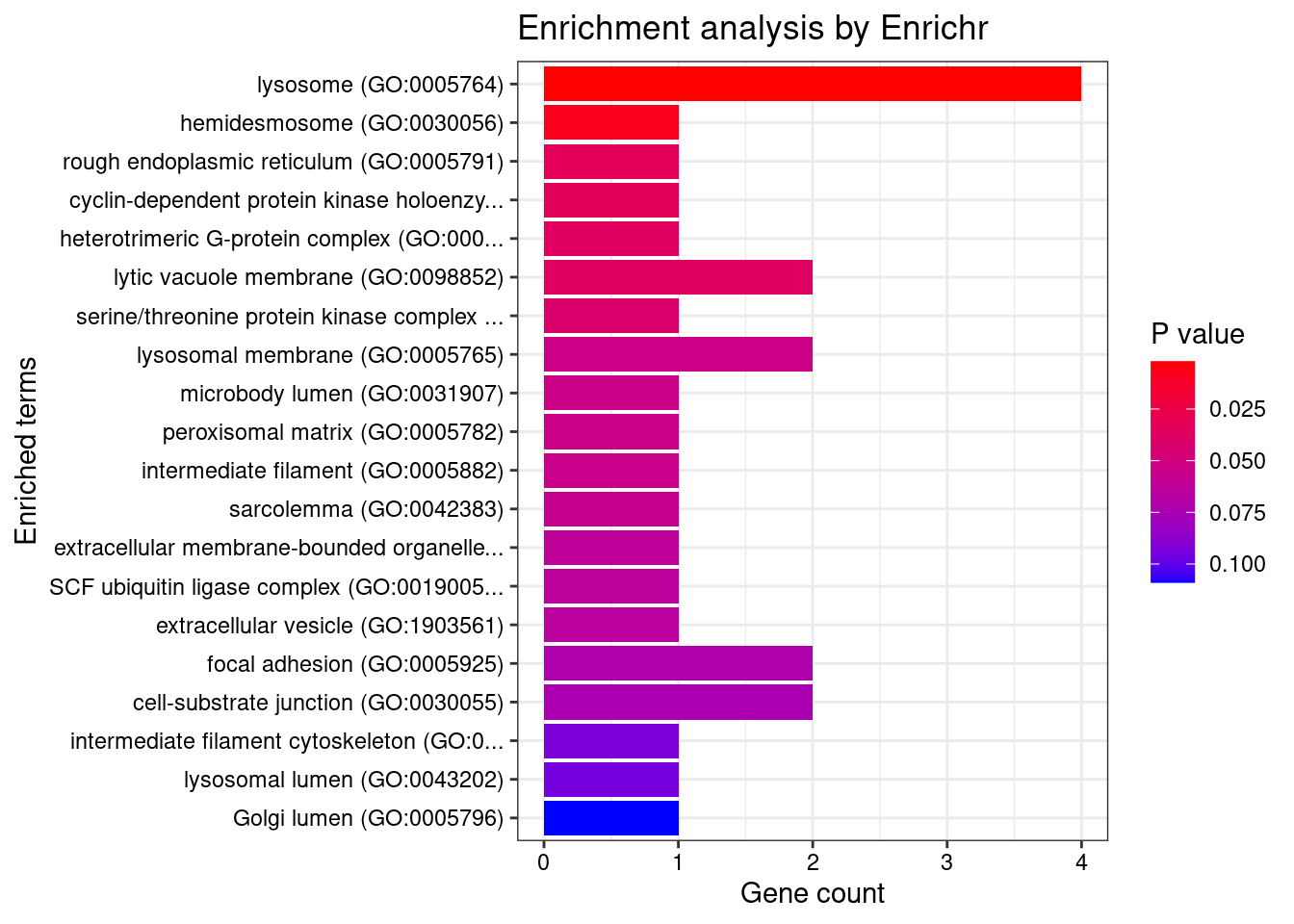
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"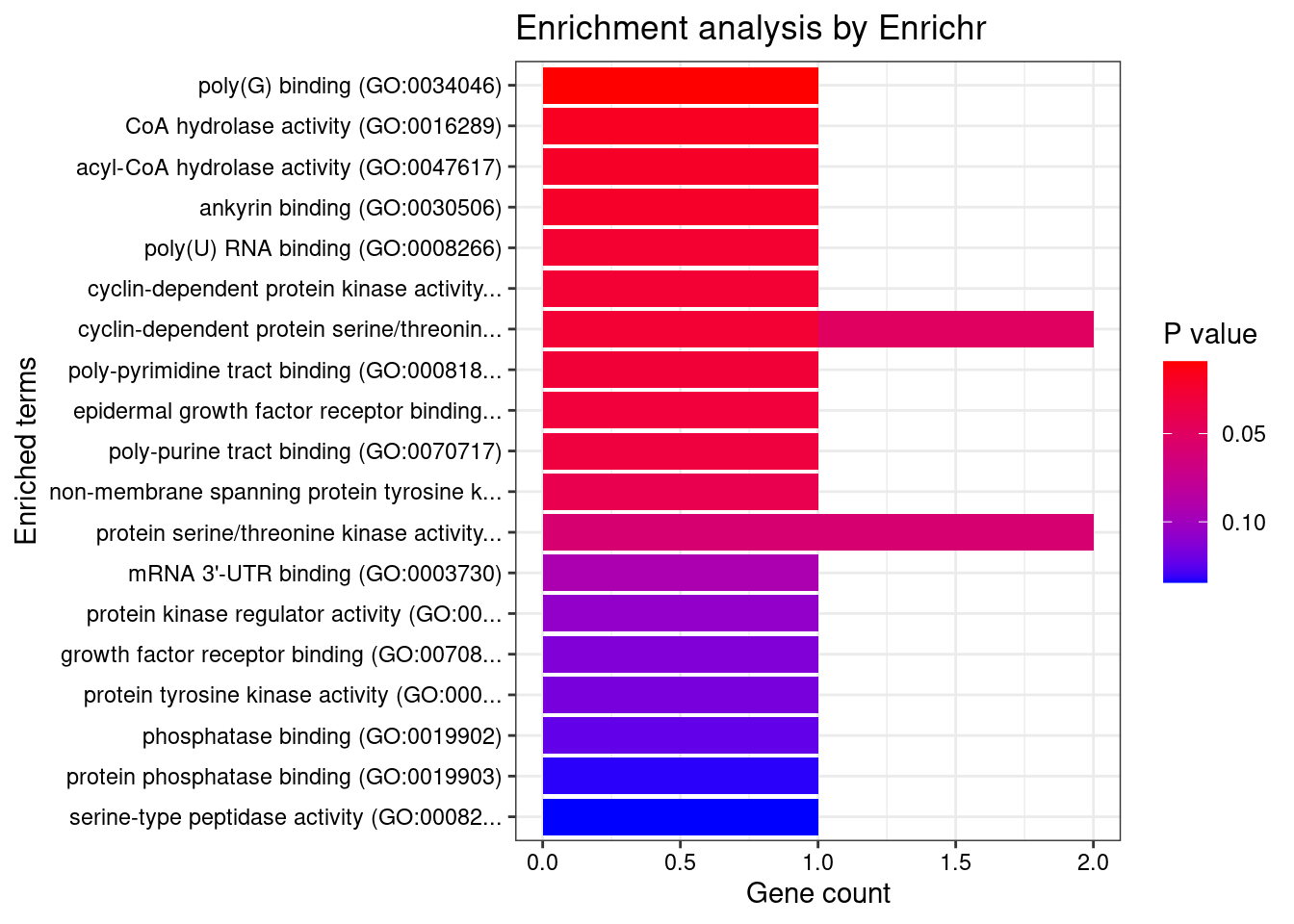
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"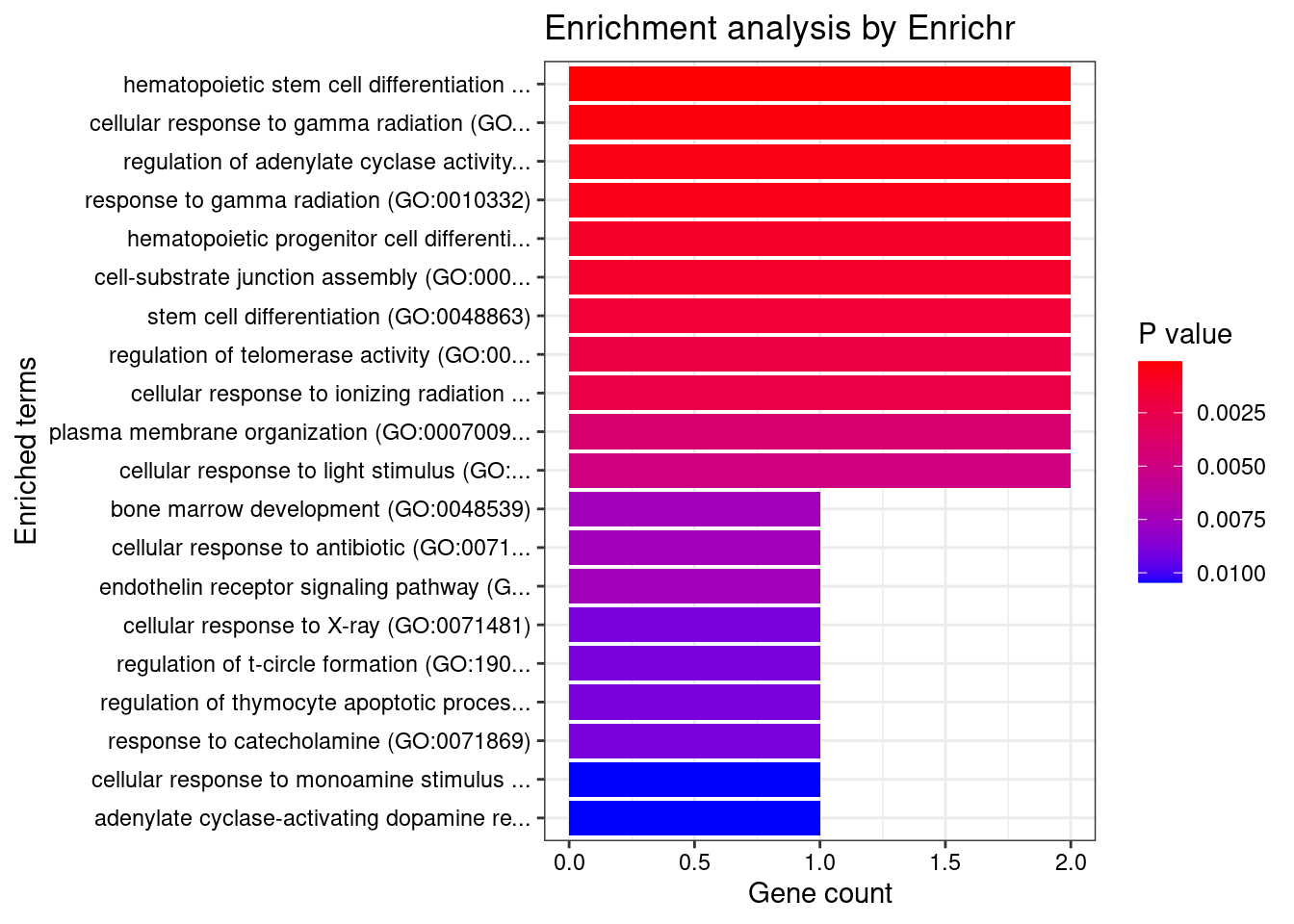
Term Overlap
1 hematopoietic stem cell differentiation (GO:0060218) 2/10
2 cellular response to gamma radiation (GO:0071480) 2/17
3 regulation of adenylate cyclase activity (GO:0045761) 2/22
4 response to gamma radiation (GO:0010332) 2/24
Adjusted.P.value Genes
1 0.03205870 HOXB4;TP53
2 0.04812932 XRCC5;TP53
3 0.04851960 EDNRB;CRHR1
4 0.04851960 XRCC5;TP53
[1] "GO_Cellular_Component_2021"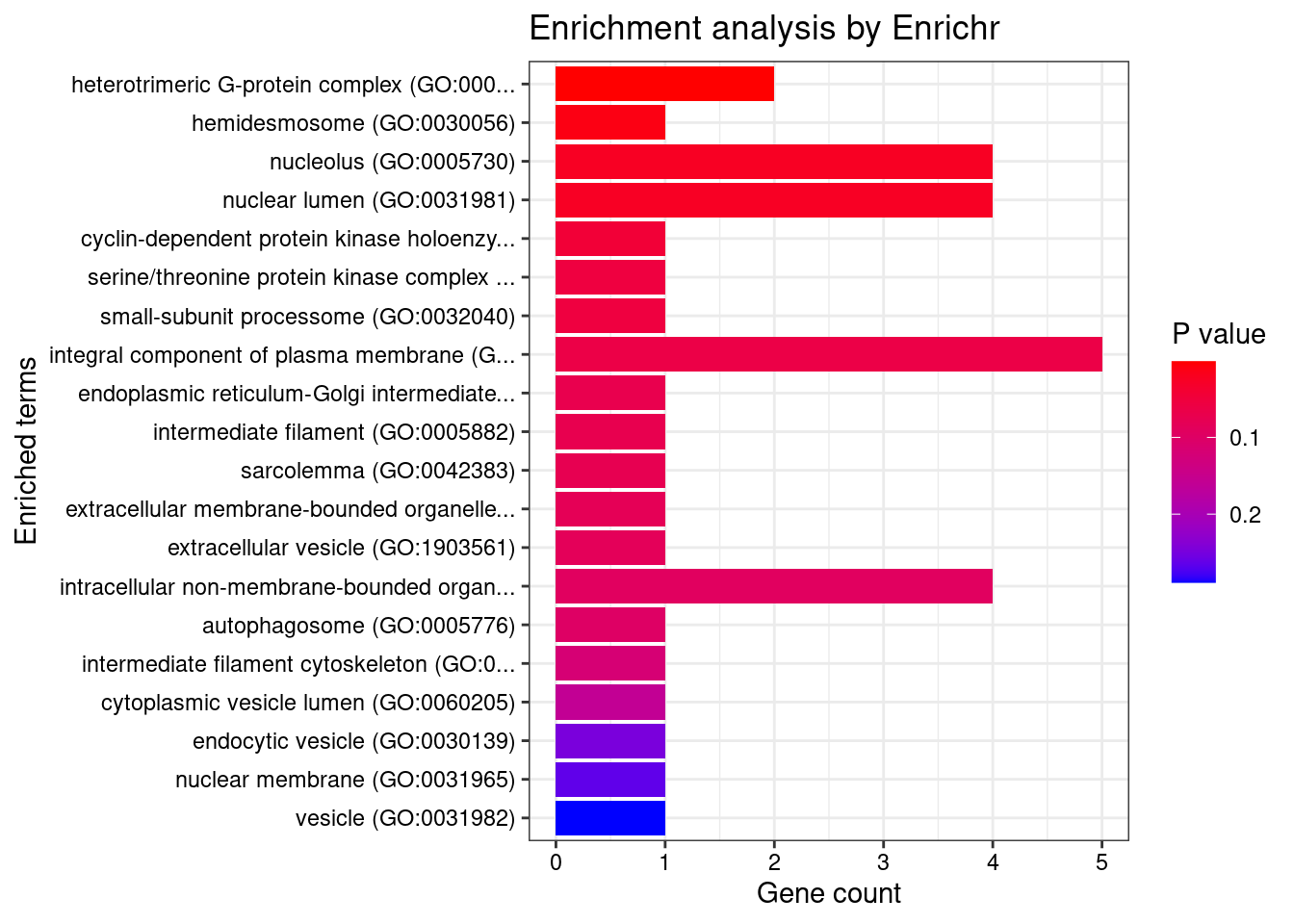
Term Overlap Adjusted.P.value
1 heterotrimeric G-protein complex (GO:0005834) 2/33 0.0390495
Genes
1 GNB1;GNG12
[1] "GO_Molecular_Function_2021"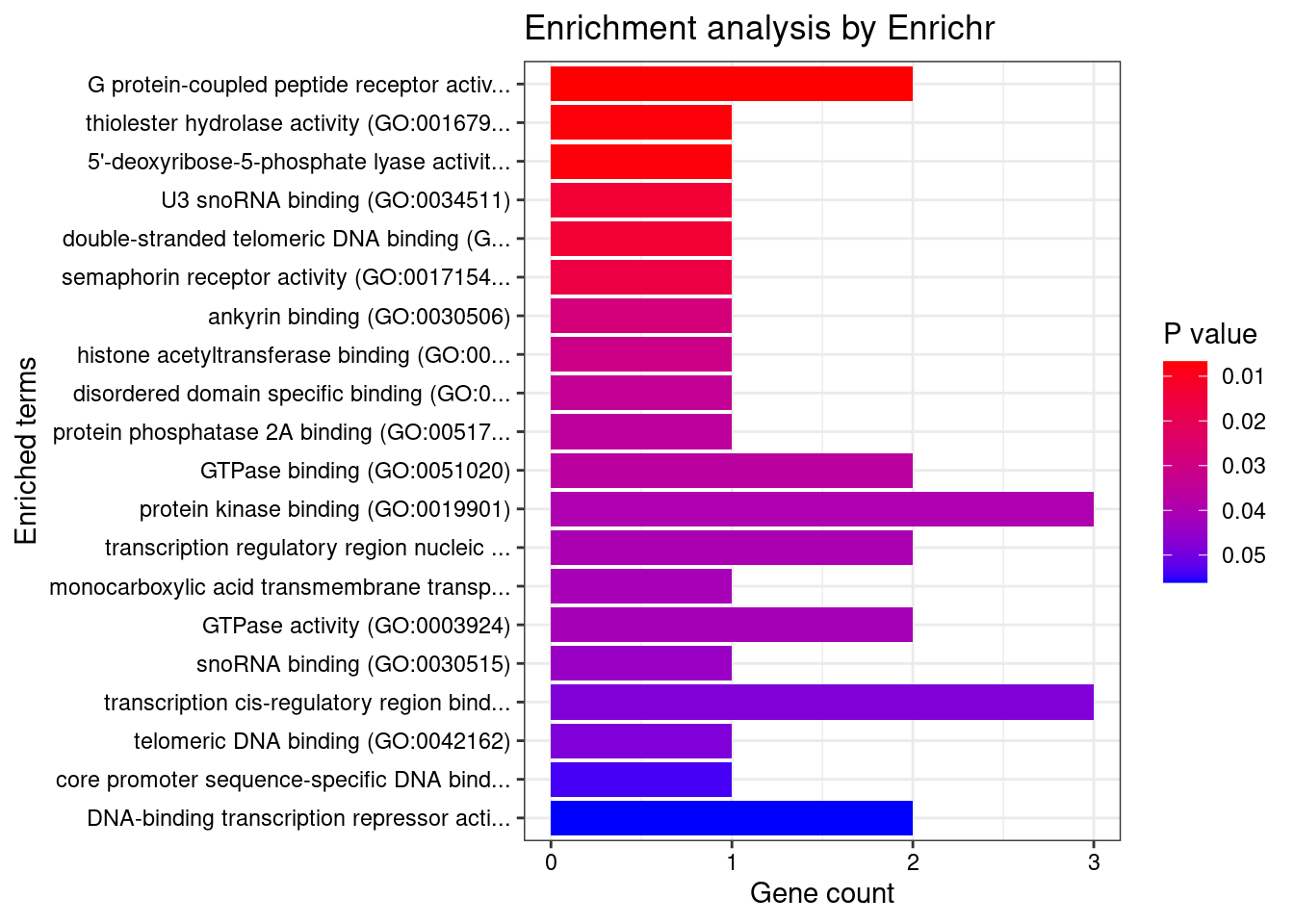
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"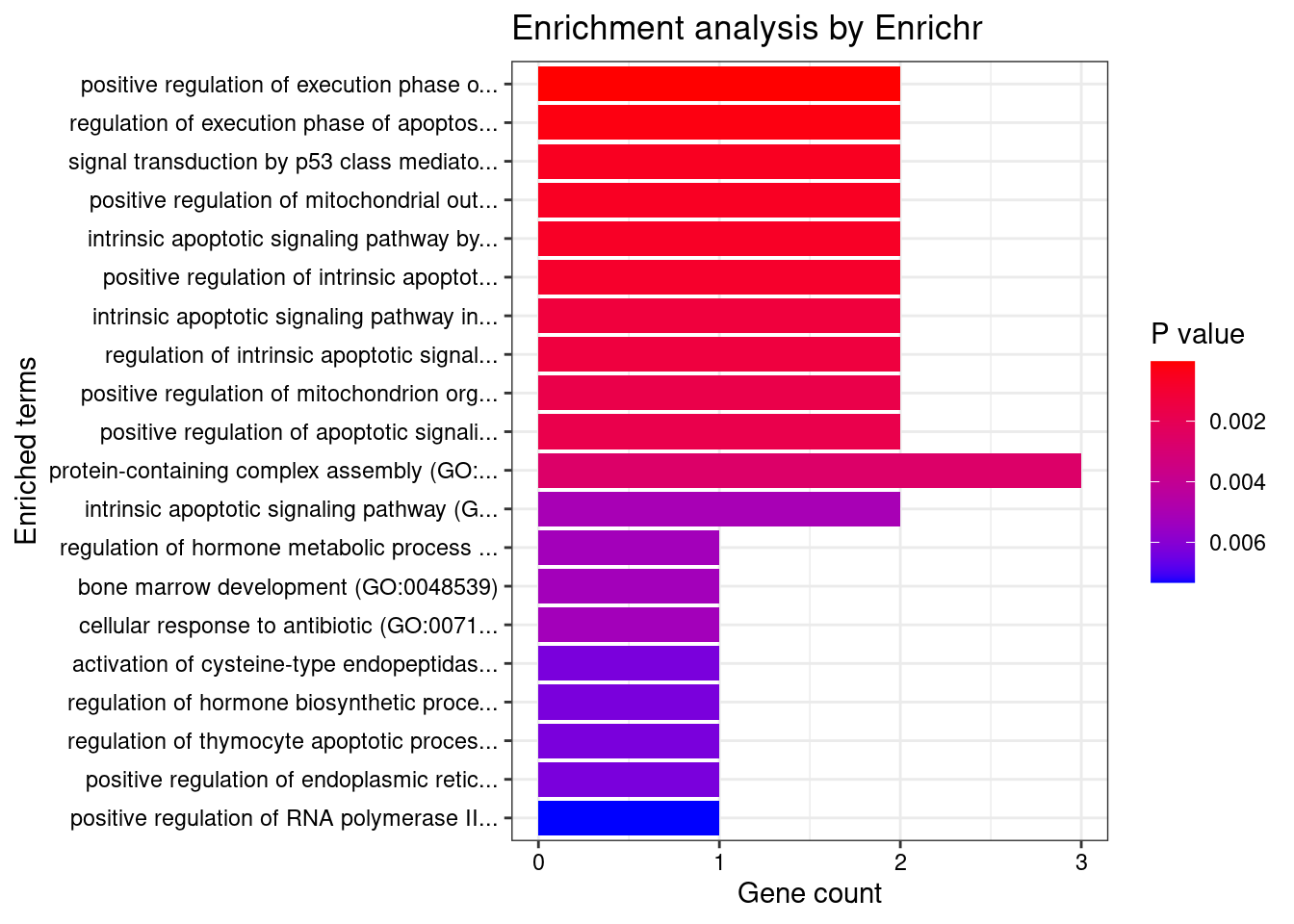
Term
1 positive regulation of execution phase of apoptosis (GO:1900119)
2 regulation of execution phase of apoptosis (GO:1900117)
3 signal transduction by p53 class mediator (GO:0072331)
4 positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway (GO:1901030)
5 intrinsic apoptotic signaling pathway by p53 class mediator (GO:0072332)
6 positive regulation of intrinsic apoptotic signaling pathway (GO:2001244)
7 intrinsic apoptotic signaling pathway in response to DNA damage (GO:0008630)
8 regulation of intrinsic apoptotic signaling pathway (GO:2001242)
9 positive regulation of mitochondrion organization (GO:0010822)
10 positive regulation of apoptotic signaling pathway (GO:2001235)
Overlap Adjusted.P.value Genes
1 2/8 0.006912061 TP53;BOK
2 2/22 0.028261114 TP53;BOK
3 2/33 0.030558564 TP53;BOK
4 2/34 0.030558564 TP53;BOK
5 2/36 0.030558564 TP53;BOK
6 2/40 0.031449036 TP53;BOK
7 2/51 0.039794497 TP53;BOK
8 2/52 0.039794497 TP53;BOK
9 2/58 0.040897644 TP53;BOK
10 2/59 0.040897644 TP53;BOK
[1] "GO_Cellular_Component_2021"
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"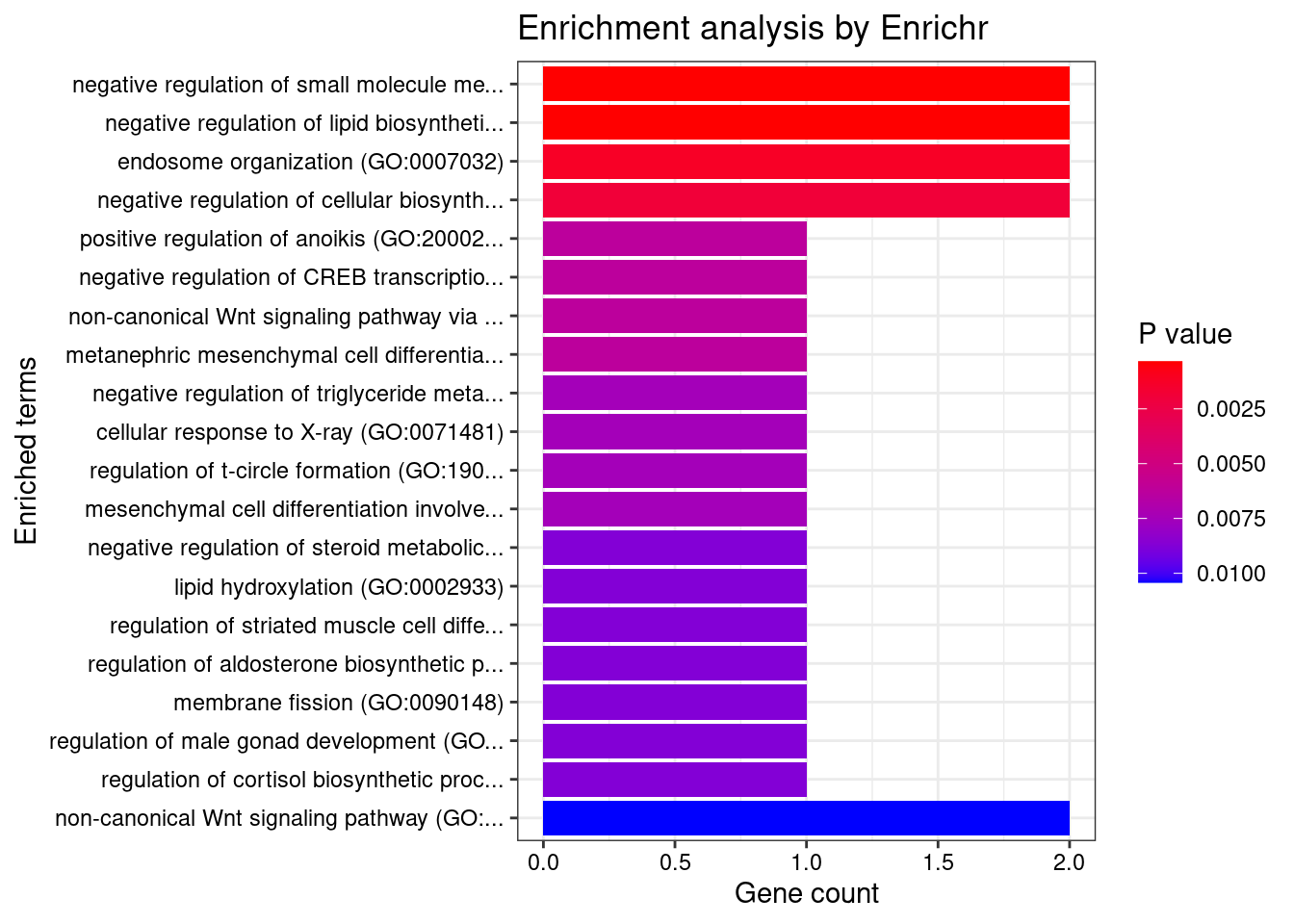
Term
1 negative regulation of small molecule metabolic process (GO:0062014)
2 negative regulation of lipid biosynthetic process (GO:0051055)
Overlap Adjusted.P.value Genes
1 2/22 0.03702394 SIK1;WNT4
2 2/22 0.03702394 SIK1;WNT4
[1] "GO_Cellular_Component_2021"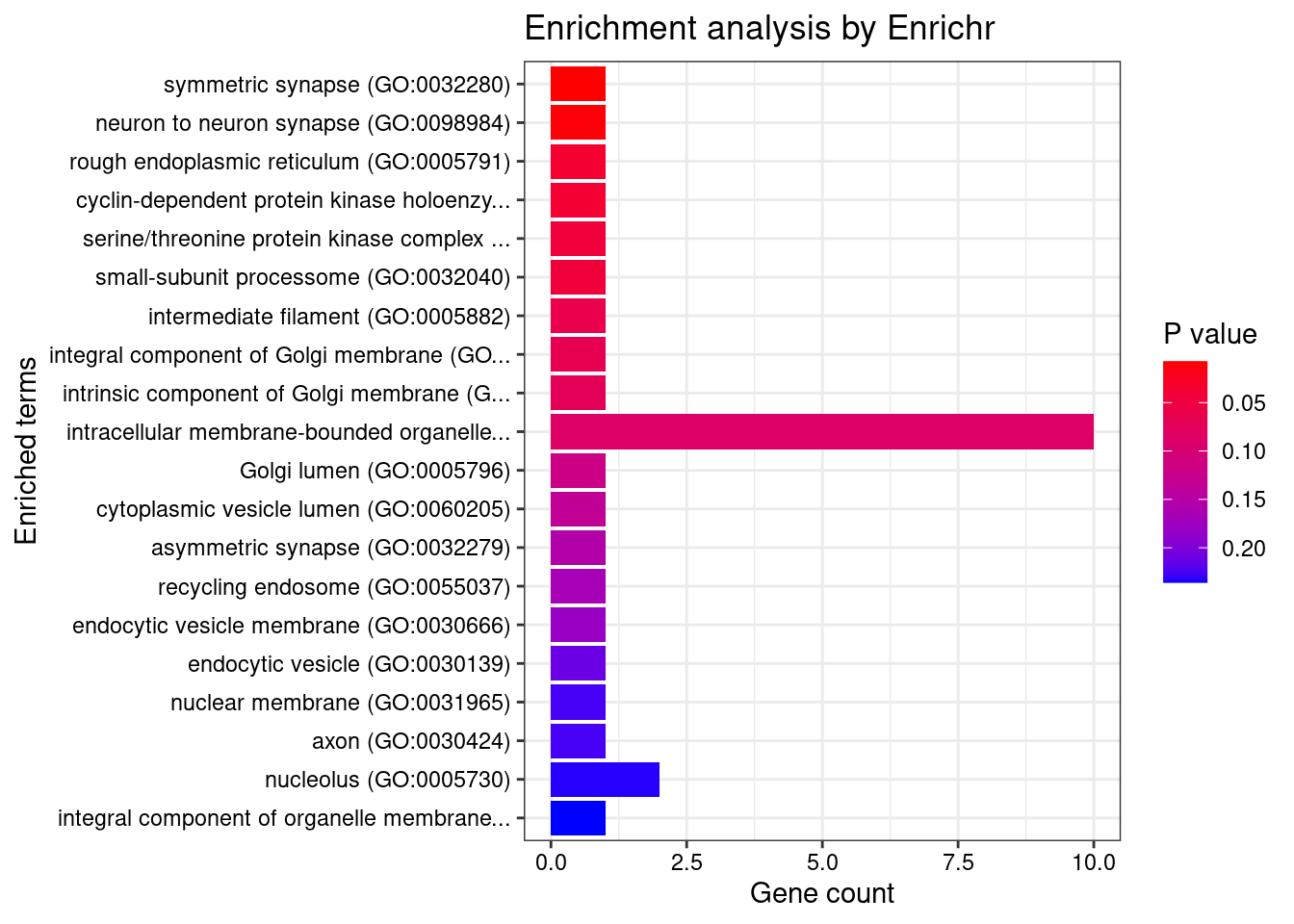
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"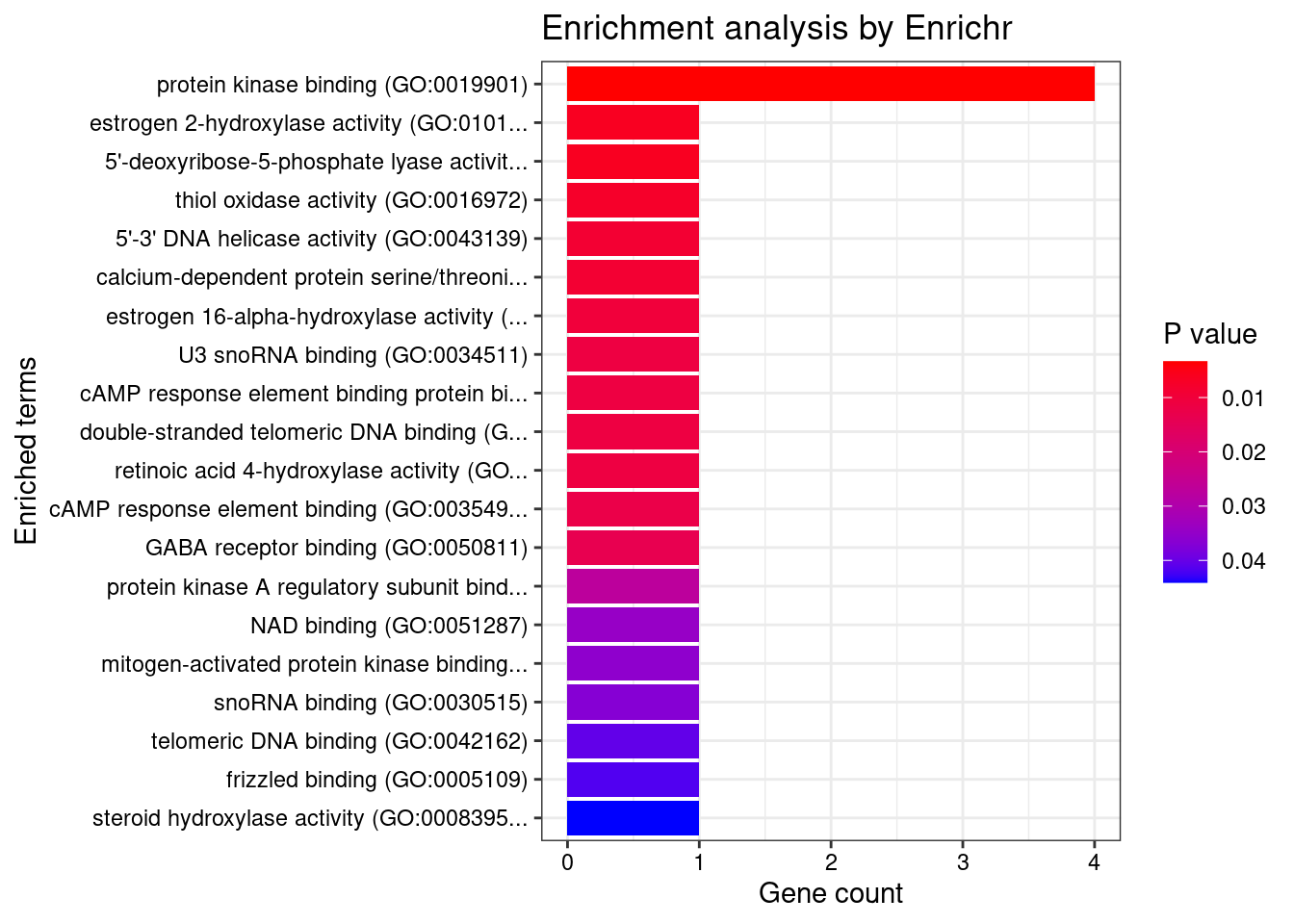
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"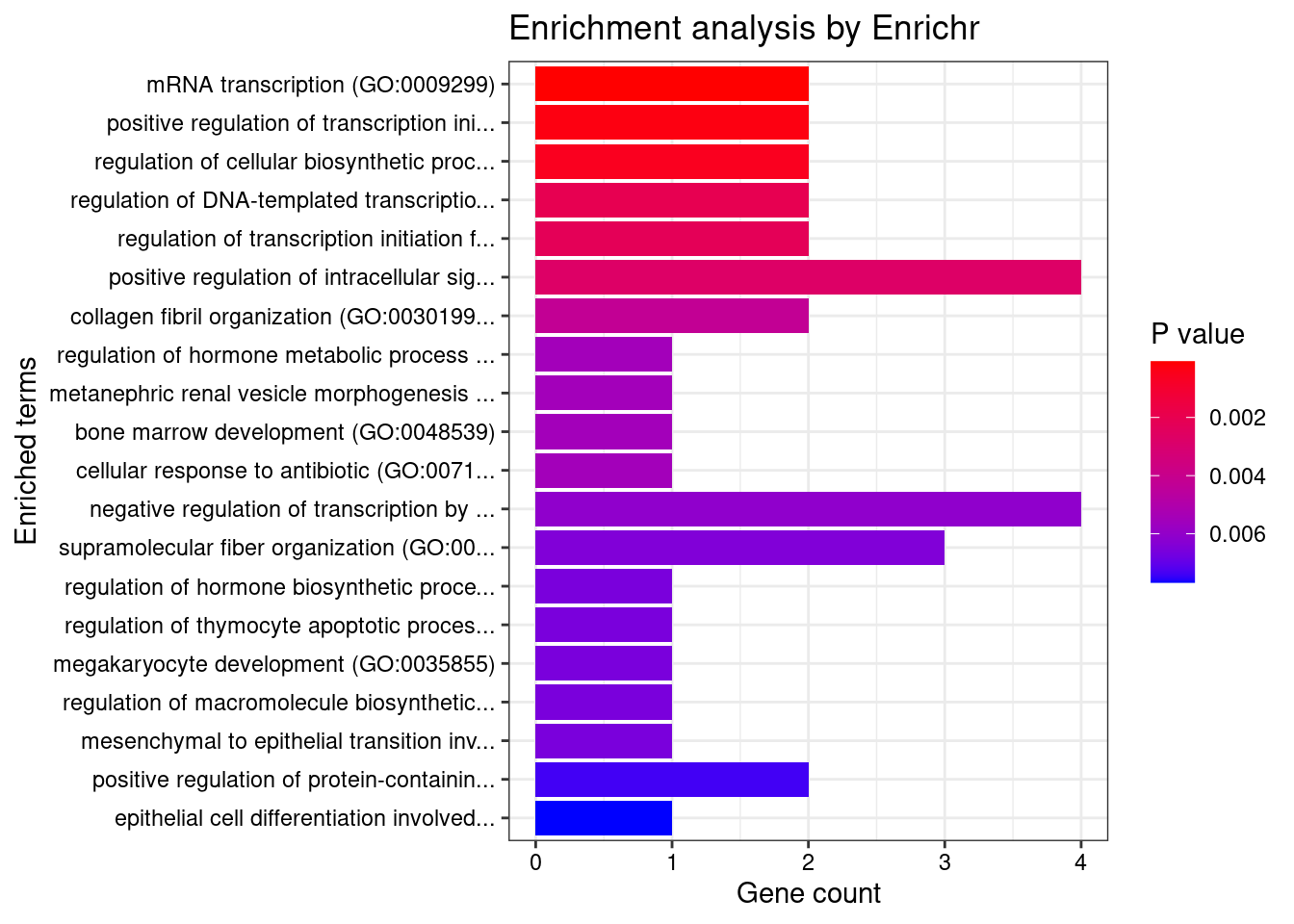
Term
1 mRNA transcription (GO:0009299)
2 positive regulation of transcription initiation from RNA polymerase II promoter (GO:0060261)
Overlap Adjusted.P.value Genes
1 2/12 0.02491316 MED1;TP53
2 2/23 0.04740195 MED1;TP53
[1] "GO_Cellular_Component_2021"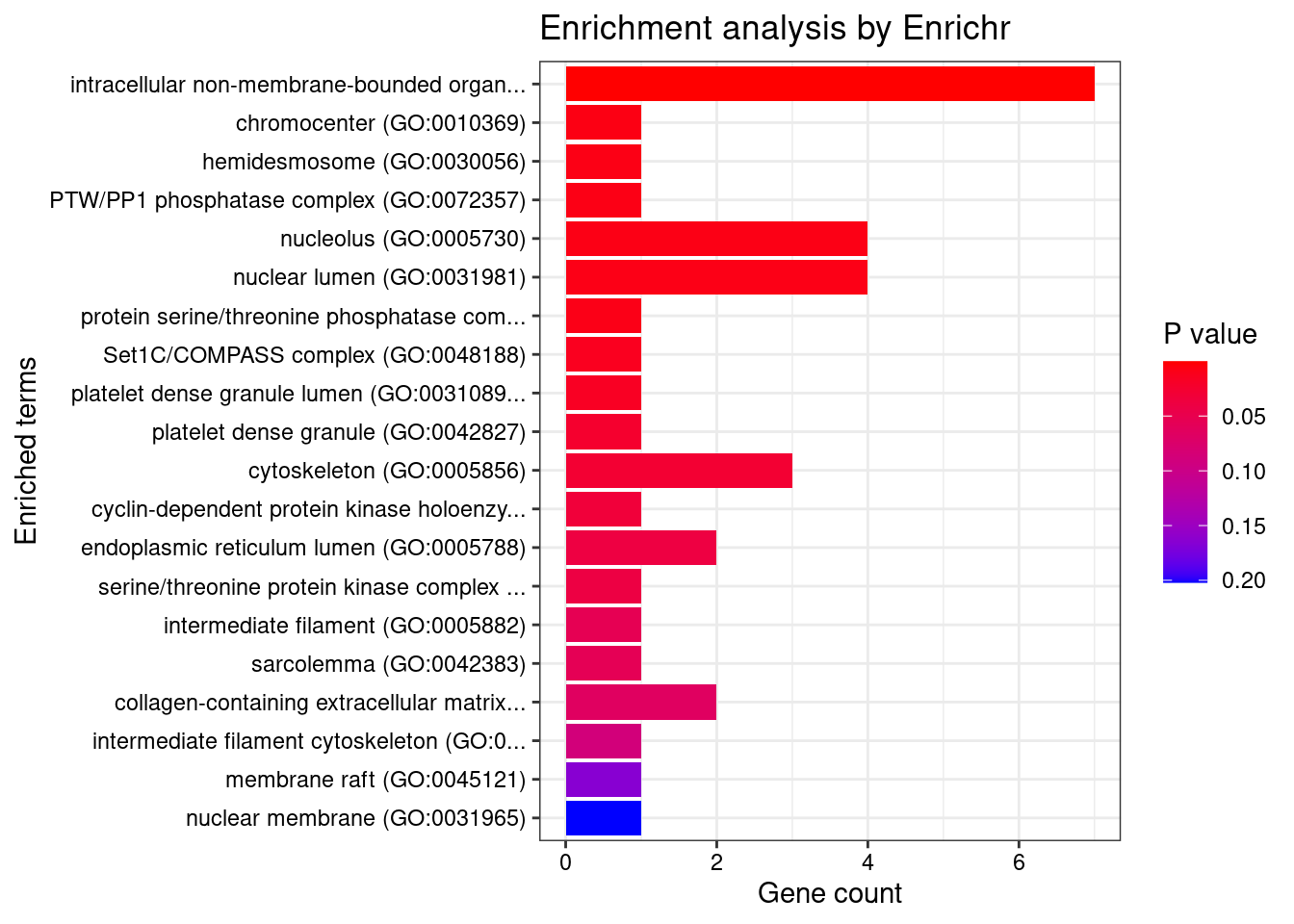
Term Overlap
1 intracellular non-membrane-bounded organelle (GO:0043232) 7/1158
2 chromocenter (GO:0010369) 1/6
3 hemidesmosome (GO:0030056) 1/7
4 PTW/PP1 phosphatase complex (GO:0072357) 1/7
5 nucleolus (GO:0005730) 4/733
6 nuclear lumen (GO:0031981) 4/745
7 protein serine/threonine phosphatase complex (GO:0008287) 1/8
Adjusted.P.value Genes
1 0.005245655 MED1;SMTN;CCND2;SALL1;WDR82;RHOC;TP53
2 0.038828066 SALL1
3 0.038828066 PLEC
4 0.038828066 WDR82
5 0.038828066 MED1;CCND2;WDR82;TP53
6 0.038828066 MED1;CCND2;WDR82;TP53
7 0.038828066 WDR82
[1] "GO_Molecular_Function_2021"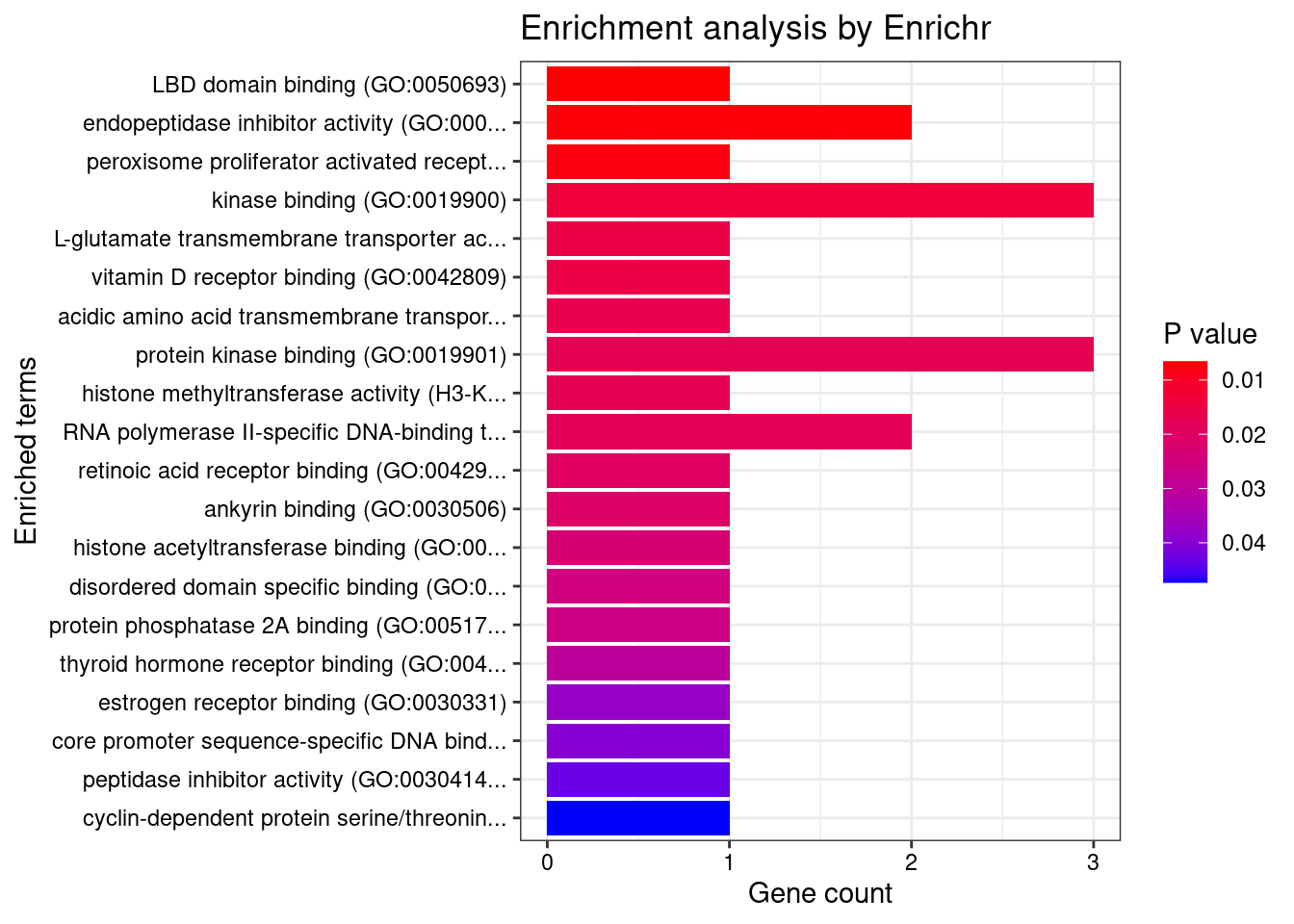
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] enrichR_3.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.6 compiler_3.6.1 pillar_1.6.1 later_0.8.0
[5] git2r_0.26.1 workflowr_1.6.2 tools_3.6.1 digest_0.6.20
[9] evaluate_0.14 lifecycle_1.0.0 tibble_3.1.2 gtable_0.3.0
[13] pkgconfig_2.0.3 rlang_0.4.11 DBI_1.1.1 curl_3.3
[17] yaml_2.2.0 xfun_0.8 httr_1.4.1 stringr_1.4.0
[21] dplyr_1.0.7 knitr_1.23 generics_0.0.2 fs_1.3.1
[25] vctrs_0.3.8 tidyselect_1.1.0 rprojroot_2.0.2 grid_3.6.1
[29] glue_1.4.2 R6_2.5.0 fansi_0.5.0 rmarkdown_1.13
[33] farver_2.1.0 purrr_0.3.4 ggplot2_3.3.3 magrittr_2.0.1
[37] whisker_0.3-2 scales_1.1.0 promises_1.0.1 htmltools_0.3.6
[41] ellipsis_0.3.2 colorspace_1.4-1 httpuv_1.5.1 labeling_0.3
[45] utf8_1.2.1 stringi_1.4.3 munsell_0.5.0 rjson_0.2.20
[49] crayon_1.4.1