Investigating MR Locus Simulation Results
wesleycrouse
2022-11-14
Last updated: 2023-06-12
Checks: 6 1
Knit directory: ctwas_applied/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210726) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d17186e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Untracked files:
Untracked: gwas.RData
Untracked: ld_R_info.RData
Untracked: z_snp_pos_ebi-a-GCST004131.RData
Untracked: z_snp_pos_ebi-a-GCST004132.RData
Untracked: z_snp_pos_ebi-a-GCST004133.RData
Untracked: z_snp_pos_scz-2018.RData
Untracked: z_snp_pos_ukb-a-360.RData
Untracked: z_snp_pos_ukb-d-30780_irnt.RData
Unstaged changes:
Modified: analysis/simulation_MRLocus.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/simulation_MRLocus.Rmd)
and HTML (docs/simulation_MRLocus.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d17186e | wesleycrouse | 2023-06-08 | updating MRLocus investigation |
| html | d17186e | wesleycrouse | 2023-06-08 | updating MRLocus investigation |
| Rmd | 7f6fbc8 | wesleycrouse | 2023-05-15 | fixing issue with plots |
| html | 7f6fbc8 | wesleycrouse | 2023-05-15 | fixing issue with plots |
| Rmd | 3e493c9 | wesleycrouse | 2023-05-15 | adding detailed MRLocus results |
| html | 3e493c9 | wesleycrouse | 2023-05-15 | adding detailed MRLocus results |
| Rmd | dba8c0e | wesleycrouse | 2023-05-14 | updating simulation plot with MRLocus |
| Rmd | c538341 | wesleycrouse | 2023-04-26 | fixing issue with ncausal plot |
MRLocus in High PVE Scenario
library(data.table)
library(mrlocus)
library(ctwas)
#load weight names
weightf <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous.pos"
weights <- as.data.frame(fread(weightf, header = T))
weights$ENSEMBL_ID <- sapply(weights$WGT, function(x){unlist(strsplit(unlist(strsplit(x,"/"))[2], "[.]"))[2]})
####################
results_df <- data.frame()
simutag_list <- c("4-1", "4-2", "4-3", "4-4", "4-5")
#simutag_list <- c("10-1", "10-2", "10-3", "10-4", "10-5")
####################
results_dir <- "/project2/mstephens/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/"
alpha <- 0.05
for (simutag in simutag_list){
load(paste0("/home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416/ukb-s80.45-adi_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes <- weights$ENSEMBL_ID[match(true_genes, weights$ID)]
n_causal <- length(true_genes)
##########
results_files <- list.files(results_dir)
results_files <- results_files[grep(".mrlocus.", results_files)]
results_files <- results_files[grep(simutag, results_files)]
results_files <- results_files[grep("result.batch", results_files)]
#results_files <- results_files[grep("result.noprune", results_files)]
results_files <- results_files[grep("_temp", results_files)]
results_files <- paste0(results_dir, results_files)
mrlocus_results <- lapply(results_files, function(x){as.data.frame(fread(x, header=T))})
mrlocus_results <- do.call(rbind, mrlocus_results)
##########
mrlocus_results$TP <- mrlocus_results$gene_id %in% true_genes
n_genes <- nrow(mrlocus_results)
n_zero_clumps <- sum(mrlocus_results$n_clumps==0)
n_one_clump <- sum(mrlocus_results$n_clumps==1)
n_sig <- sum(sign(mrlocus_results$CI_10)==sign(mrlocus_results$CI_90), na.rm=T)
n_sig_twoplus <- sum(sign(mrlocus_results$CI_10)==sign(mrlocus_results$CI_90) & mrlocus_results$n_clumps>1, na.rm=T)
n_true_positive <- sum(sign(mrlocus_results$CI_10)==sign(mrlocus_results$CI_90) & mrlocus_results$TP, na.rm=T)
n_causal_oneplus_clump <- sum(mrlocus_results$n_clumps>0 & mrlocus_results$TP, na.rm=T)
n_causal_twoplus_clumps <- sum(mrlocus_results$n_clumps>1 & mrlocus_results$TP, na.rm=T)
n_tp_twoplus <- sum(sign(mrlocus_results$CI_10)==sign(mrlocus_results$CI_90) & mrlocus_results$TP & mrlocus_results$n_clumps>1, na.rm=T)
results_current <- data.frame(simutag=simutag,
n_causal=n_causal,
n_genes=n_genes,
n_zero_clumps=n_zero_clumps,
n_one_clump=n_one_clump,
n_sig=n_sig,
n_causal_oneplus_clump=n_causal_oneplus_clump,
n_causal_twoplus_clumps=n_causal_twoplus_clumps,
n_true_positive=n_true_positive,
n_tp_twoplus=n_tp_twoplus,
n_sig_twoplus=n_sig_twoplus)
results_df <- rbind(results_df, results_current)
}
results_df$percent_tp <- results_df$n_true_positive/results_df$n_sig
####################
#summary of MRLocus results for each simulation
results_df simutag n_causal n_genes n_zero_clumps n_one_clump n_sig
1 4-1 106 8192 1662 2884 275
2 4-2 105 8192 1681 2905 288
3 4-3 136 8192 1669 2876 331
4 4-4 132 8192 1673 2893 308
5 4-5 123 8192 1738 2860 335
n_causal_oneplus_clump n_causal_twoplus_clumps n_true_positive n_tp_twoplus
1 81 49 28 13
2 89 53 24 10
3 98 47 37 16
4 105 51 31 12
5 100 55 42 19
n_sig_twoplus percent_tp
1 118 0.10181818
2 108 0.08333333
3 133 0.11178248
4 136 0.10064935
5 131 0.12537313#averaging over simulations
colMeans(results_df[,colnames(results_df)[2:10]]) n_causal n_genes n_zero_clumps
120.4 8192.0 1684.6
n_one_clump n_sig n_causal_oneplus_clump
2883.6 307.4 94.6
n_causal_twoplus_clumps n_true_positive n_tp_twoplus
51.0 32.4 14.0 Investigating results for a single simulation
#load weight names
weightf <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous.pos"
weights <- as.data.frame(fread(weightf, header = T))
weights$ENSEMBL_ID <- sapply(weights$WGT, function(x){unlist(strsplit(unlist(strsplit(x,"/"))[2], "[.]"))[2]})
####################
#results_dir <- "/project2/mstephens/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/"
simutag <- "4-1"
####################
#load true positive genes and SNPs from simulation
load(paste0("/home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416/ukb-s80.45-adi_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes <- weights$ENSEMBL_ID[match(true_genes, weights$ID)]
n_causal <- length(true_genes)
true_gene_effects <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$e.beta}))
names(true_gene_effects) <- true_genes
true_snps <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cSNP}))
true_snp_effects <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$s.theta}))
names(true_snp_effects) <- true_snps
####################
#load MRLocus results
results_files <- list.files(results_dir)
results_files <- results_files[grep(".mrlocus.result.batch", results_files)]
results_files <- results_files[grep(simutag, results_files)]
results_files <- results_files[grep("_temp", results_files)]
results_files <- paste0(results_dir, results_files)
mrlocus_results <- lapply(results_files, function(x){as.data.frame(fread(x, header=T))})
mrlocus_results <- do.call(rbind, mrlocus_results)
#load the TWAS z scores
twas <- paste0("/project2/mstephens/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb-s80.45-adi_simu", simutag, ".Adipose_Subcutaneous.coloc.result")
twas <- fread(twas, header = T)
twas$gene_id <- weights$ENSEMBL_ID[match(twas$ID, weights$ID)]
mrlocus_results$twas_p <- twas$TWAS.P[match(mrlocus_results$gene_id, twas$gene_id)]
#denote significant genes
alpha <- 0.05
sig_thresh_twas <- alpha/nrow(weights)
mrlocus_results$causal <- mrlocus_results$gene_id %in% true_genes
mrlocus_results$sig <- sign(mrlocus_results$CI_10)==sign(mrlocus_results$CI_90)
#drop results for genes that did not run in MRLocus
mrlocus_results <- mrlocus_results[!is.na(mrlocus_results$sig),]
#drop results for genes that had <2 clumps
mrlocus_results <- mrlocus_results[mrlocus_results$n_clumps>1,]
#drop results that did not run in TWAS
mrlocus_results <- mrlocus_results[!is.na(mrlocus_results$twas_p),]
####################
#add cTWAS results
ctwas <- as.data.frame(data.table::fread("/project2/mstephens/causalTWAS/simulations/simulation_ctwas_rss_20210416/ukb-s80.45-adi_simu4-1_config1.susieIrss.txt"))
ctwas <- ctwas[ctwas$type=="gene",]
ctwas$gene_id <- twas$gene_id[match(ctwas$id, twas$ID)]
mrlocus_results$ctwas_pip <- ctwas$susie_pip[match(mrlocus_results$gene_id, ctwas$gene_id)]
####################
head(mrlocus_results[order(mrlocus_results$twas_p),], 20) chromosome gene_id n_clumps alpha CI_10 CI_90
4907 7 ENSG00000243335 2 -0.81257860 -1.73396451 -0.023893658
5401 5 ENSG00000072364 3 0.68700162 0.40504926 0.982889623
4727 7 ENSG00000237310 2 -0.22902827 -0.40057137 -0.058073354
1590 5 ENSG00000164574 3 0.02016277 -0.04403925 0.085984948
6339 7 ENSG00000272568 2 -0.29833993 -0.51897487 -0.081279172
4377 7 ENSG00000229180 2 -0.12730656 -0.46592700 0.211618536
2004 7 ENSG00000169919 2 -0.20679783 -0.52218491 0.074665067
4001 6 ENSG00000215022 2 0.10704806 -0.08477306 0.295991576
1576 6 ENSG00000164385 2 0.24743372 0.10893124 0.387811428
1583 6 ENSG00000164465 2 0.19345806 0.05752564 0.349509401
1030 1 ENSG00000153898 2 0.21065411 0.02960322 0.391463748
1675 12 ENSG00000165714 2 -0.29891763 -0.56694783 -0.037006074
2157 3 ENSG00000172215 3 -0.18929235 -0.34650084 -0.034868698
1064 7 ENSG00000154710 3 -0.05042623 -0.08437666 -0.018401842
3089 2 ENSG00000184898 2 -0.35777072 -0.77628551 0.049058198
82 13 ENSG00000134874 2 -0.42808047 -1.00227600 0.155620487
265 11 ENSG00000137700 2 0.19164374 -0.08438019 0.488912649
7808 2 ENSG00000123609 2 -0.16050952 -0.38335106 0.054014761
3167 11 ENSG00000186166 3 0.15281020 -0.11921504 0.432461292
4615 7 ENSG00000234585 3 -0.02188476 -0.04405970 0.001151294
twas_p causal sig ctwas_pip
4907 9.72e-33 FALSE TRUE 0.004551816
5401 1.07e-30 TRUE TRUE 0.548505024
4727 6.76e-30 FALSE TRUE 0.002209191
1590 4.09e-28 FALSE FALSE 0.021123908
6339 2.03e-26 TRUE TRUE 0.999992919
4377 1.00e-25 FALSE FALSE 0.002905230
2004 6.32e-24 FALSE FALSE 0.001692352
4001 9.21e-23 TRUE FALSE 0.999999943
1576 6.85e-21 TRUE TRUE 0.999994211
1583 9.67e-20 FALSE TRUE 0.606859551
1030 7.41e-19 TRUE TRUE 0.448164858
1675 1.64e-18 TRUE TRUE 0.999265697
2157 1.48e-17 FALSE TRUE 0.478867838
1064 1.08e-16 FALSE TRUE 0.001624743
3089 9.83e-16 TRUE FALSE 0.621690999
82 1.24e-15 FALSE FALSE 0.009461171
265 9.07e-15 TRUE FALSE 0.820883824
7808 2.25e-14 FALSE FALSE 0.018403307
3167 9.47e-13 FALSE FALSE 0.017768348
4615 1.11e-11 FALSE FALSE 0.044163645ld_R_dir = "/project2/mstephens/causalTWAS/ukbiobank/ukb_LDR_s80.45/s80.45.2_LDR"
gwas <- "/project2/mstephens/causalTWAS/simulations/simulation_ctwas_rss_20210416/ukb-s80.45-adi_simu4-1.snpgwas.txt.gz"
eqtl <- "/project2/mstephens/causalTWAS/GTEx_v7_all/Adipose_Subcutaneous.allpairs_processed.txt.gz"
weightf <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous.pos"
outputdir <- "/home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/"
outname <- "mrlocus_report_temp"
####################
ld_bedfs <- list.files(outputdir, pattern="*.bed")
ld_bedfs <- data.frame(chr=sapply(ld_bedfs, function(x){as.numeric(unlist(strsplit(unlist(strsplit(x, "_"))[2], "chr"))[2])}),
file=ld_bedfs)
rownames(ld_bedfs) <- NULL
ld_bedfs <- ld_bedfs[order(ld_bedfs$chr),]
ld_bedfs$file <- paste0(outputdir, tools::file_path_sans_ext(ld_bedfs$file))
temp_dir <- paste0(outputdir, "temp_mrlocus/")
outname_base <- rev(unlist(strsplit(outname, "/")))[1]
####################
#define function for harmonization
harmonize_sumstat_ld <- function(sumstats, ldref, effect_var="z"){
snpnames <- intersect(sumstats$id, ldref$id)
if (length(snpnames) != 0){
ss.idx <- which(sumstats$id %in% snpnames)
ld.idx <- match(sumstats$id[ss.idx], ldref$id)
qc <- ctwas:::allele.qc(sumstats$alt[ss.idx], sumstats$ref[ss.idx],
ldref$alt[ld.idx], ldref$ref[ld.idx])
ifflip <- qc[["flip"]]
ifremove <- !qc[["keep"]]
flip.idx <- ss.idx[ifflip]
sumstats[flip.idx, c("alt", "ref")] <- sumstats[flip.idx, c("ref", "alt")]
sumstats[flip.idx, effect_var] <- -sumstats[flip.idx, effect_var]
remove.idx <- ss.idx[ifremove]
if (length(remove.idx) != 0) {
sumstats <- sumstats[-remove.idx,,drop = F]
}
}
return(sumstats)
}
####################
#load GWAS
gwas <- as.data.frame(fread(gwas, header = T))
#format GWAS data
gwas <- gwas[,!(colnames(gwas) %in% c("t.value", "PVALUE"))]
#drop variants that are non uniquely identified by ID
gwas <- gwas[!(gwas$id %in% gwas$id[duplicated(gwas$id)]),]
####################
#load weight names
weights <- as.data.frame(fread(weightf, header = T))
weights$ENSEMBL_ID <- sapply(weights$WGT, function(x){unlist(strsplit(unlist(strsplit(x,"/"))[2], "[.]"))[2]})
####################
#load eQTL; in GTEx, the eQTL effect allele is the ALT allele
eqtl <- as.data.frame(fread(eqtl, header = T))
#trim version number from ensembl IDs
eqtl_gene_id_crosswalk <- unique(eqtl$gene_id)
eqtl_gene_id_crosswalk <- data.frame(original=eqtl_gene_id_crosswalk,
trimmed=sapply(eqtl_gene_id_crosswalk, function(x){unlist(strsplit(x, "[.]"))[1]}))
eqtl$gene_id <- eqtl_gene_id_crosswalk[eqtl$gene_id, "trimmed"]
eqtl <- eqtl[, c("rs_id_dbSNP147_GRCh37p13", "gene_id", "gene_name", "ref", "alt", "slope", "slope_se", "pval_nominal")]
eqtl <- dplyr::rename(eqtl, id="rs_id_dbSNP147_GRCh37p13")
#drop genes without FUSION weights
eqtl <- eqtl[eqtl$gene_id %in% weights$ENSEMBL_ID,]
#drop entries not uniquely identified by gene_id and variant id (variant not biallelic)
eqtl_unique_id_gene <- paste0(eqtl$id, eqtl$gene_id)
eqtl <- eqtl[!(eqtl_unique_id_gene %in% eqtl_unique_id_gene[duplicated(eqtl_unique_id_gene)]),]
rm(eqtl_unique_id_gene)
####################
#LD files
ld_R_files <- paste0(ld_R_dir, "/", list.files(ld_R_dir))
ld_R_files <- ld_R_files[grep(".RDS", ld_R_files)]
ld_R_info_files <- paste0(ld_R_dir, "/", list.files(ld_R_dir))
ld_R_info_files <- ld_R_info_files[grep(".Rvar", ld_R_info_files)]
ld_R_info <- lapply(1:length(ld_R_info_files), function(x){y <- fread(ld_R_info_files[x]); y$index <- x; as.data.frame(y)})
ld_R_info <- do.call(rbind, ld_R_info)
####################
#subset to SNPs in all three datasets
snplist <- intersect(intersect(gwas$id, ld_R_info$id), eqtl$id)
gwas <- gwas[gwas$id %in% snplist,]
eqtl <- eqtl[eqtl$id %in% snplist,]
ld_R_info <- ld_R_info[ld_R_info$id %in% snplist,]
rm(snplist)
####################
#harmonize GWAS to LD reference
gwas <- harmonize_sumstat_ld(gwas, ld_R_info, effect_var="Estimate")
#harmonize eQTL to LD reference
eqtl <- harmonize_sumstat_ld(eqtl, ld_R_info, effect_var="slope")
####################
#load gene location information
# gtf <- rtracklayer::import("/project2/mstephens/causalTWAS/GTEx_v7_all/gencode.v19.genes.v7.patched_contigs.gtf")
# gtf_df <- as.data.frame(gtf)
# save(gtf_df, file="/project2/mstephens/causalTWAS/GTEx_v7_all/gencode.v19.genes.v7.patched_contigs.RData")
load("/project2/mstephens/causalTWAS/GTEx_v7_all/gencode.v19.genes.v7.patched_contigs.RData")
#keep only gene-level information
gtf_df <- gtf_df[gtf_df$type=="gene",]
#trim ensembl ID version number
gtf_df$gene_id <- sapply(gtf_df$gene_id, function(x){unlist(strsplit(x, "[.]"))[1]})
#drop genes without FUSION weights
gtf_df <- gtf_df[gtf_df$gene_id %in% weights$ENSEMBL_ID,]
####################
#subset to SNPs in all three datasets again - catch discordant variants between GWAS and eQTL after harmonization
snplist <- intersect(intersect(gwas$id, ld_R_info$id), eqtl$id)
gwas <- gwas[gwas$id %in% snplist,]
eqtl <- eqtl[eqtl$id %in% snplist,]
ld_R_info <- ld_R_info[ld_R_info$id %in% snplist,]
rm(snplist)
####################
ldref_files <- readLines("/project2/mstephens/causalTWAS/ukbiobank/ukb_pgen_s80.45/ukb-s80.45.2_pgenfs.txt")
ld_pvarfs <- sapply(ldref_files, prep_pvar, outputdir = temp_dir)
save.image("/home/wcrouse/scratch-midway2/mrlocus_image.RData")True negatives
ENSG00000164574
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000164574"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr5_s80.45.2.FUSION5 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000164574.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000164574.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr5_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:22:27 INFO::ENSG00000164574par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)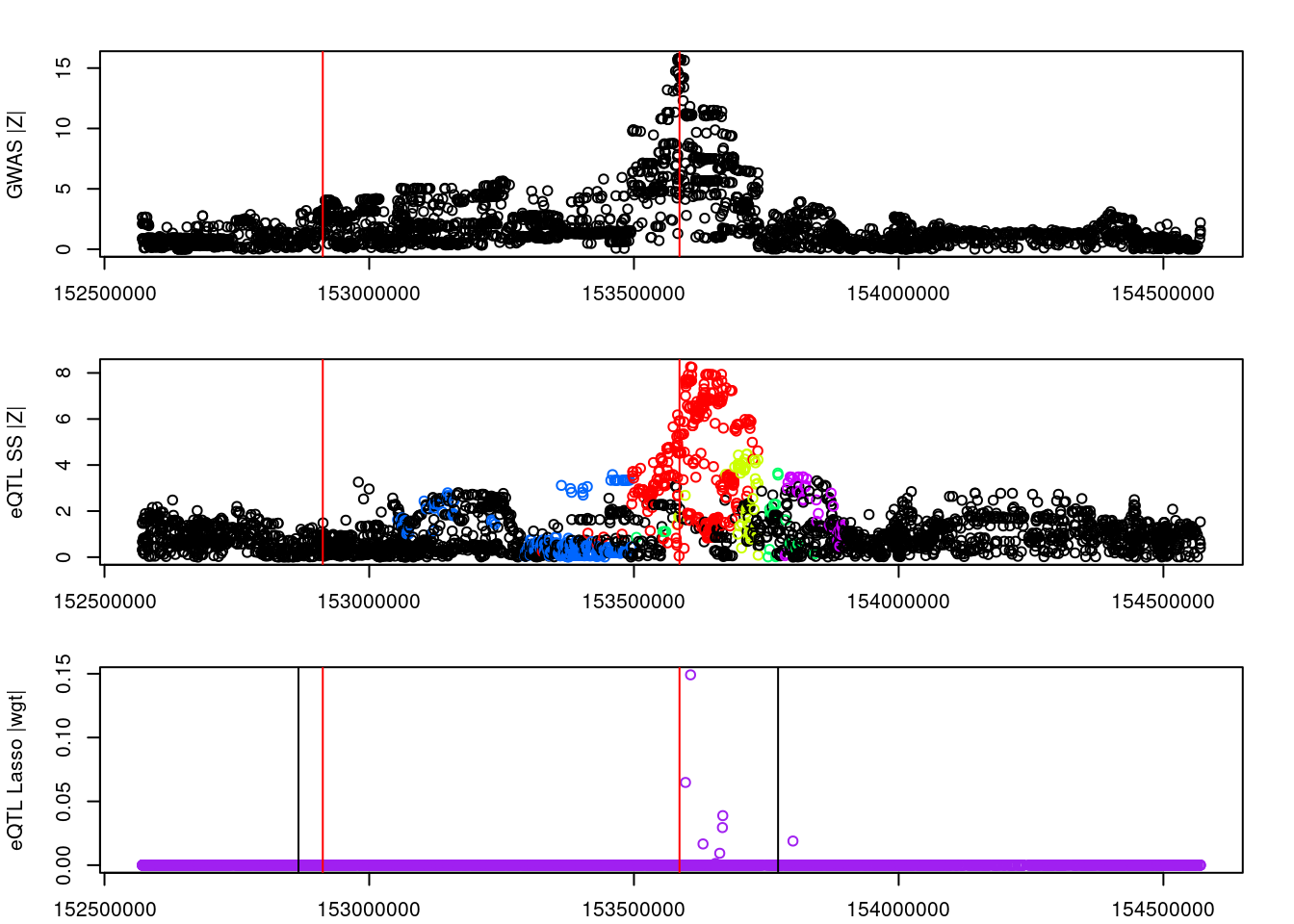
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 391,53,25,236,42post: 60,17,12,16,15data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)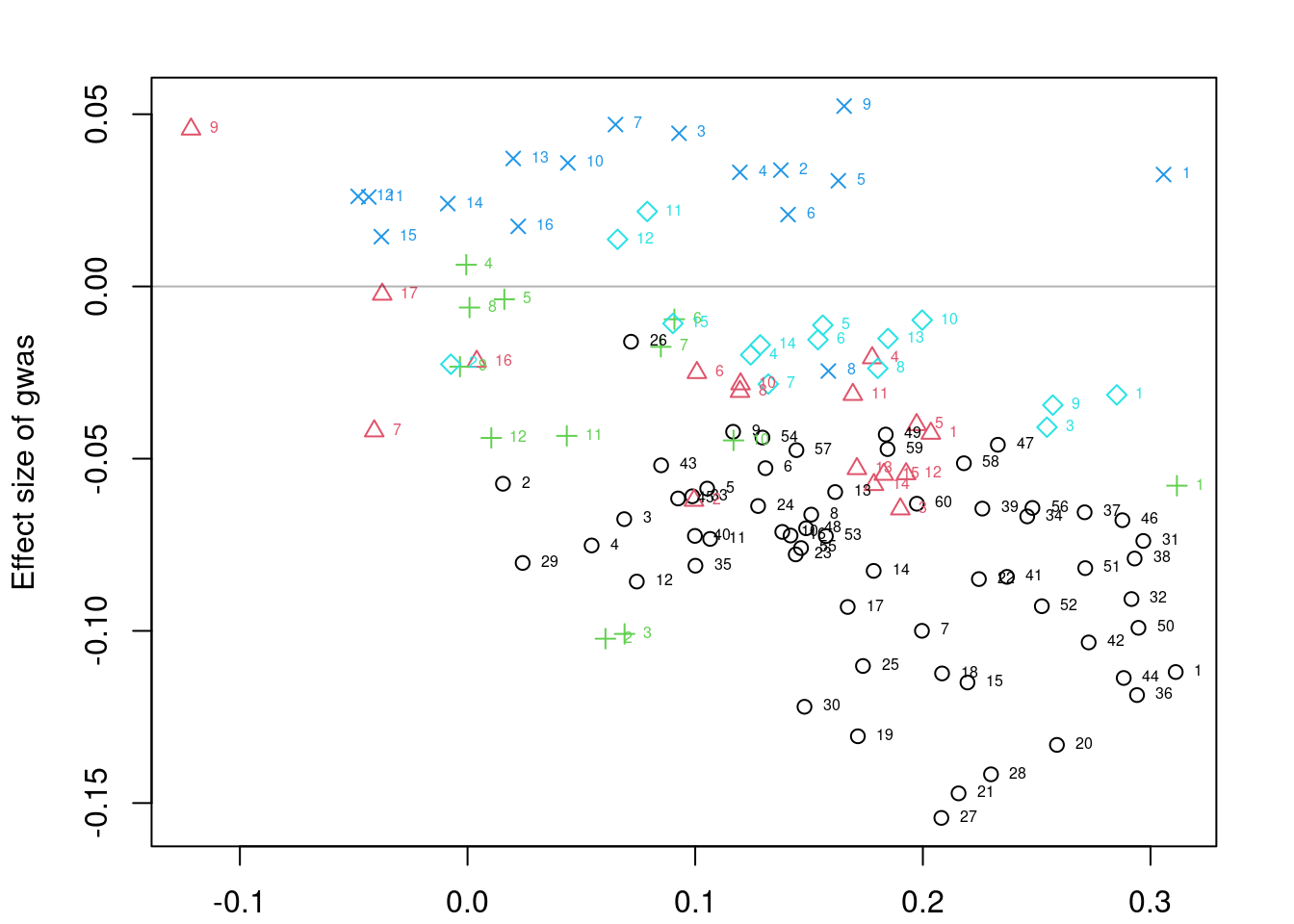
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 1837 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 1329 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.68, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 370 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 1106 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.29, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 556 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 999 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.07, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 796 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)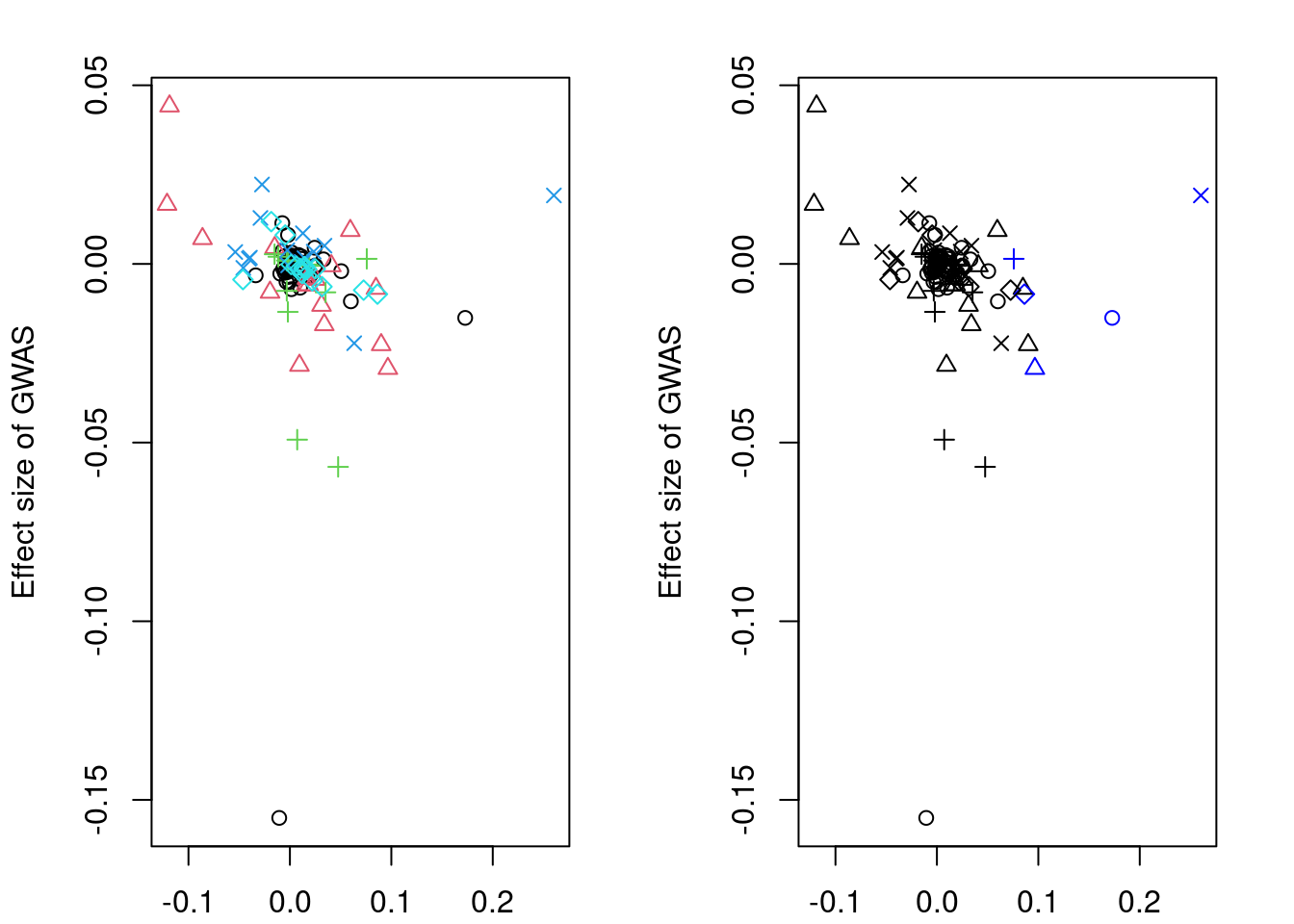
res$beta_hat_a
[1] 0.17282472 0.09660027 0.07579629 0.26026197 0.08630545
$beta_hat_b
[1] -0.015097863 -0.029275199 0.001431053 0.019172375 -0.008445798
$sd_a
[1] 0.0376575 0.0708576 0.0853399 0.0853019 0.0818514
$sd_b
[1] 0.009967241 0.015663260 0.021558256 0.021714154 0.018479744
$alleles
id ref eff
44 rs17553077 C T
63 rs7737824 A G
78 rs11746823 G T
90 rs816007 G A
106 rs113811083 C T#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:24:36 INFO::ENSG00000164574par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)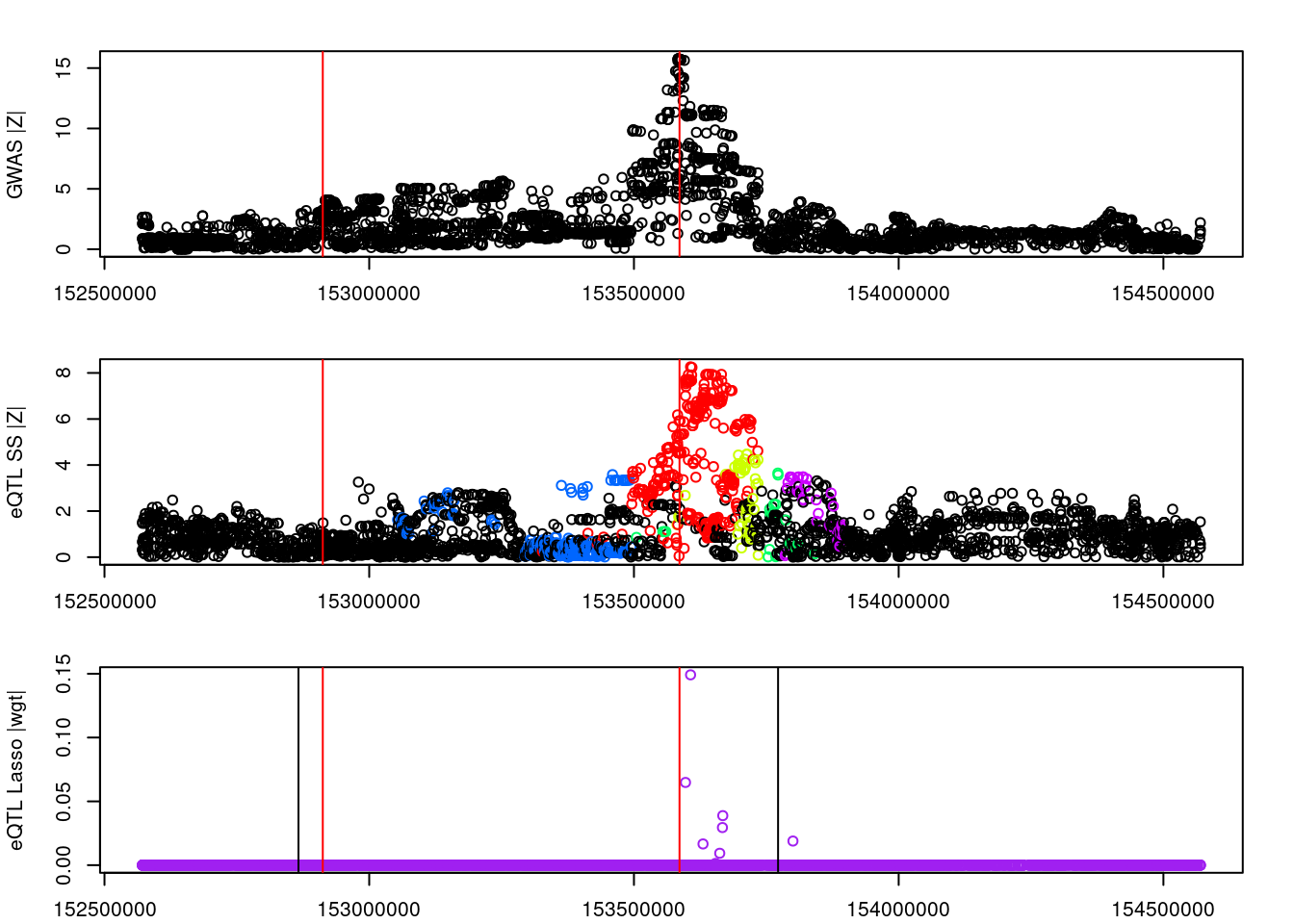
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
4549809 rs17553077 7.653907 5 153665854 -11.405619 FALSE 0 1
4549223 rs816007 3.584141 5 153459514 1.496219 FALSE 0 4
4549544 rs7737824 2.682846 5 153596813 -4.126465 FALSE 0 2
4550146 rs113811083 -3.484155 5 153794979 1.703955 FALSE 0 5
4550092 rs11746823 3.651199 5 153771825 -2.683634 FALSE 0 3
trim color
4549809 FALSE #FF0000
4549223 FALSE #0066FF
4549544 TRUE #CCFF00
4550146 TRUE #CC00FF
4550092 TRUE #00FF66par(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])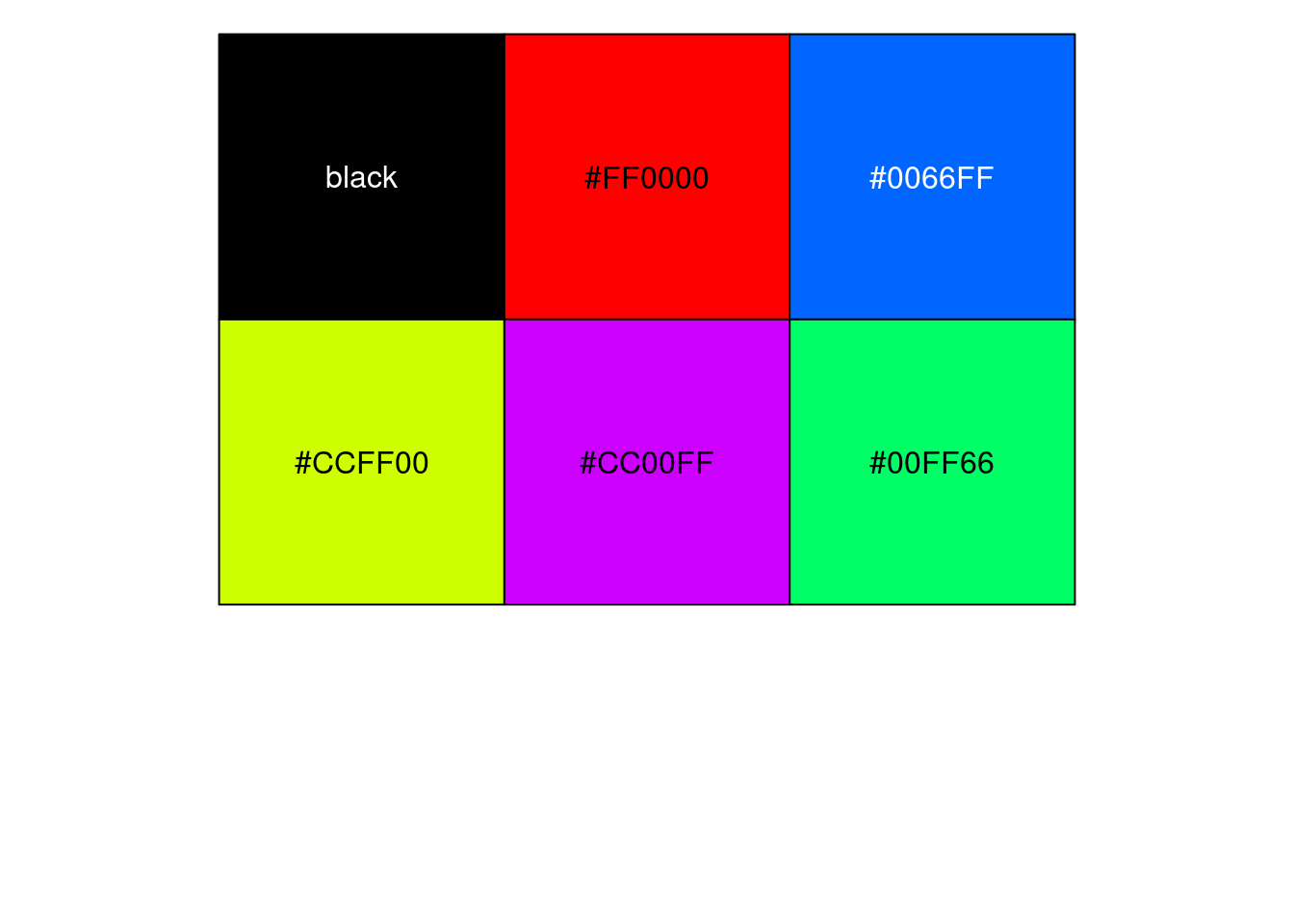
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}Warning: There were 1146 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemspar(mfrow=c(1,1))
#display results
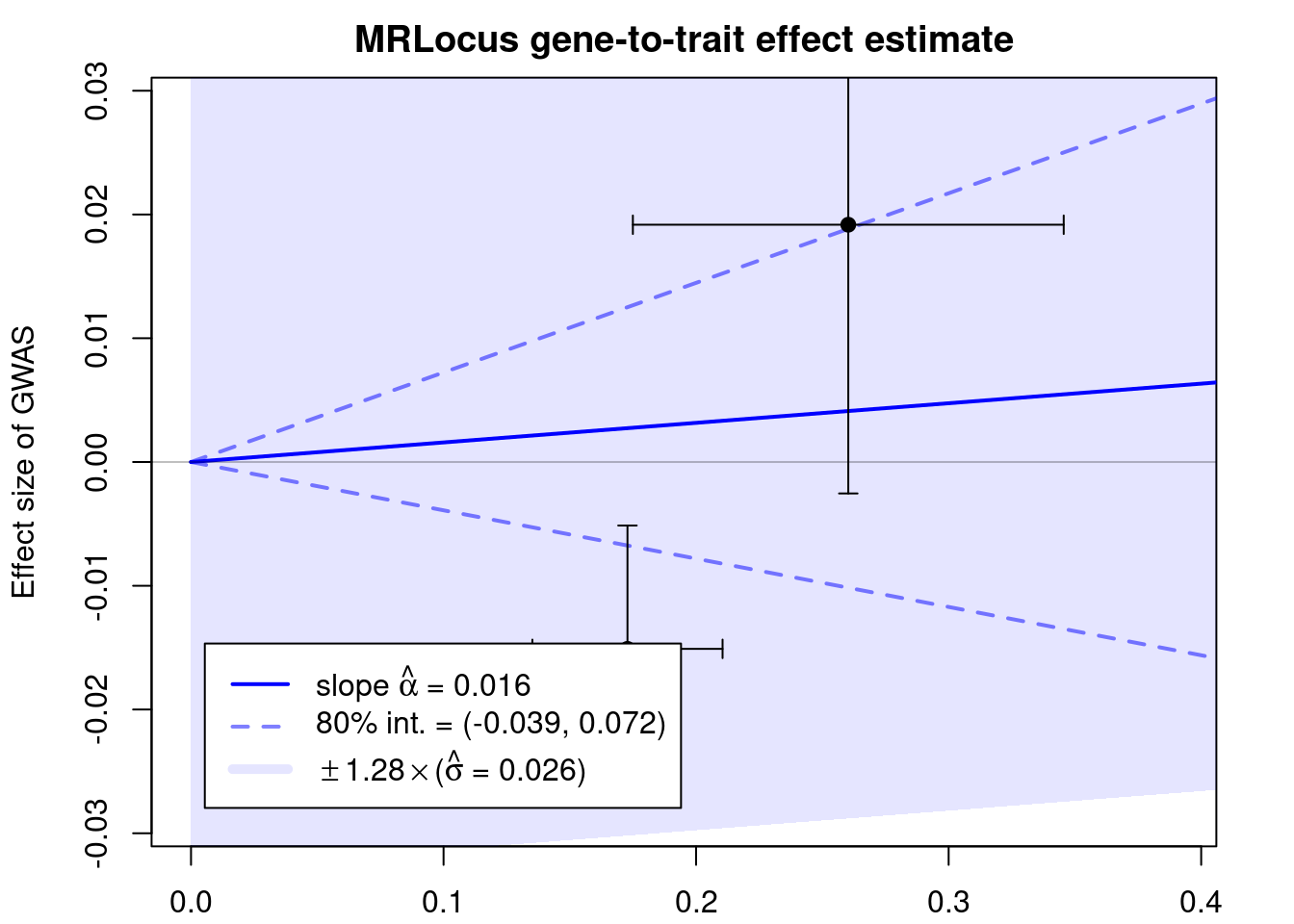
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)Inference for Stan model: slope.
4 chains, each with iter=10000; warmup=5000; thin=1;
post-warmup draws per chain=5000, total post-warmup draws=20000.
mean se_mean sd 10% 90% n_eff Rhat
alpha 0.016 0 0.043 -0.039 0.072 7699 1.000
sigma 0.026 0 0.019 0.006 0.052 6145 1.001
Samples were drawn using NUTS(diag_e) at Mon Jun 12 15:24:38 2023.
For each parameter, n_eff is a crude measure of effective sample size,
and Rhat is the potential scale reduction factor on split chains (at
convergence, Rhat=1).if (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
True positives
ENSG00000072364
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000072364"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr5_s80.45.2.FUSION5 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000072364.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000072364.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr5_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:25:24 INFO::ENSG00000072364par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)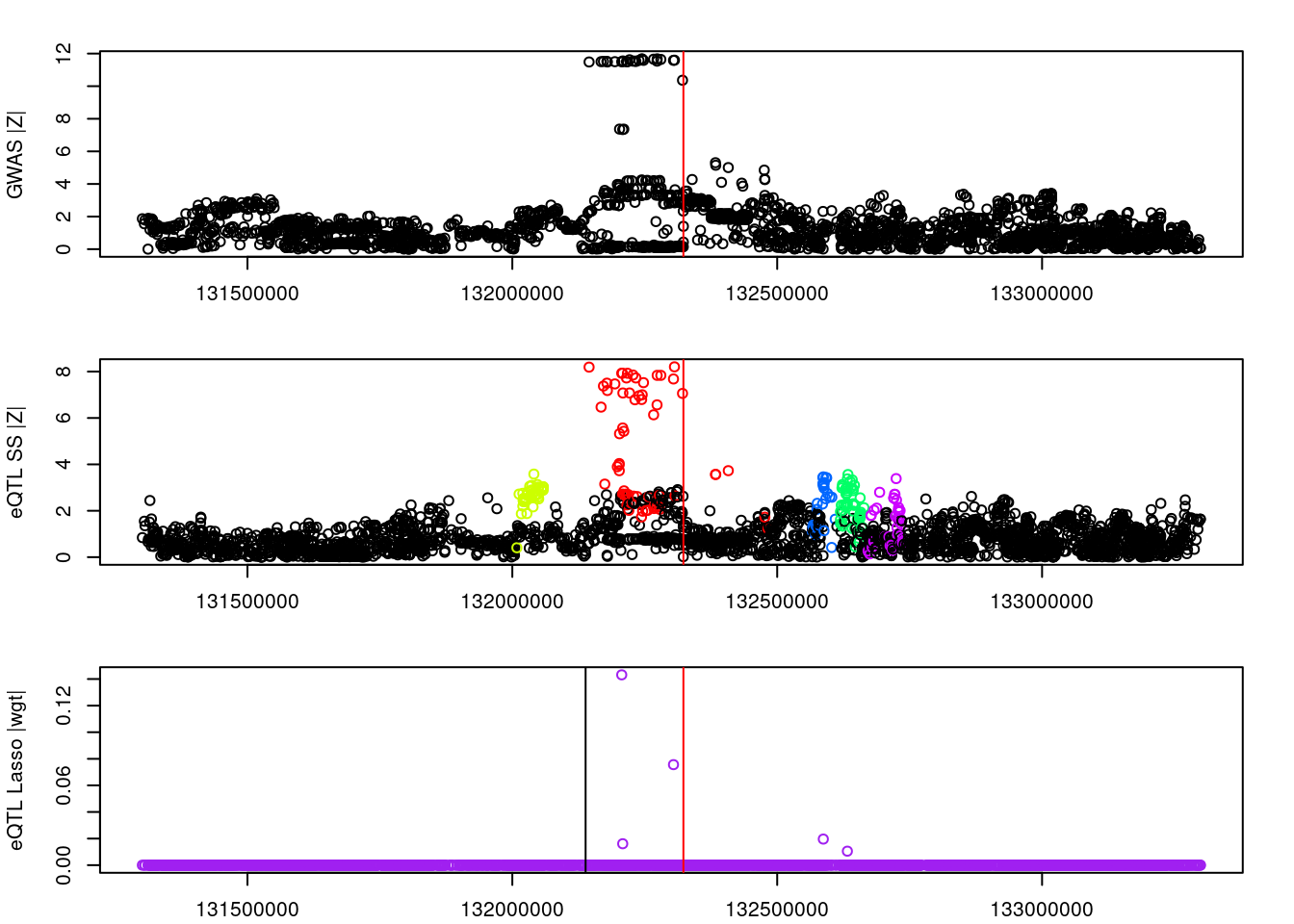
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 63,44,62,27,42post: 10,06,27,17,22data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 785 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.24, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 890 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.32, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 1652 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 579 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.61, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 701 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 45 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 1 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.05, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 856 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 144 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.08, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)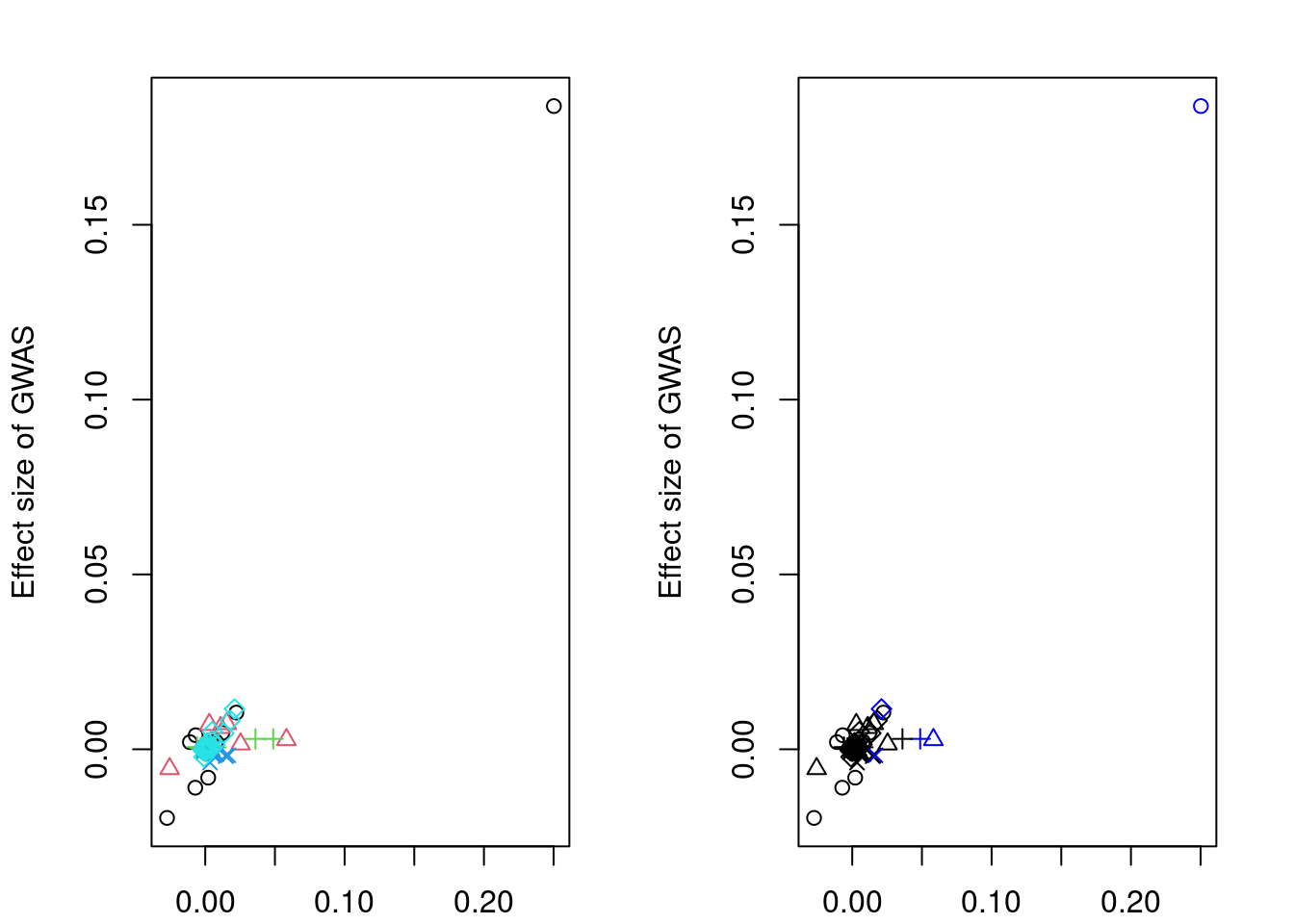
res$beta_hat_a
[1] 0.25018837 0.05824561 0.04875937 0.01657236 0.02099882
$beta_hat_b
[1] 0.183939946 0.002743535 0.002980109 -0.001716928 0.011542343
$sd_a
[1] 0.0346275 0.0342004 0.0251199 0.0239703 0.0344152
$sd_b
[1] 0.01768557 0.01568073 0.01124450 0.01034667 0.01403960
$alleles
id ref eff
1 rs11951542 A G
11 rs10069772 G A
17 rs6893561 C T
57 rs1560086 G A
74 rs11738464 C T#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:26:33 INFO::ENSG00000072364par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)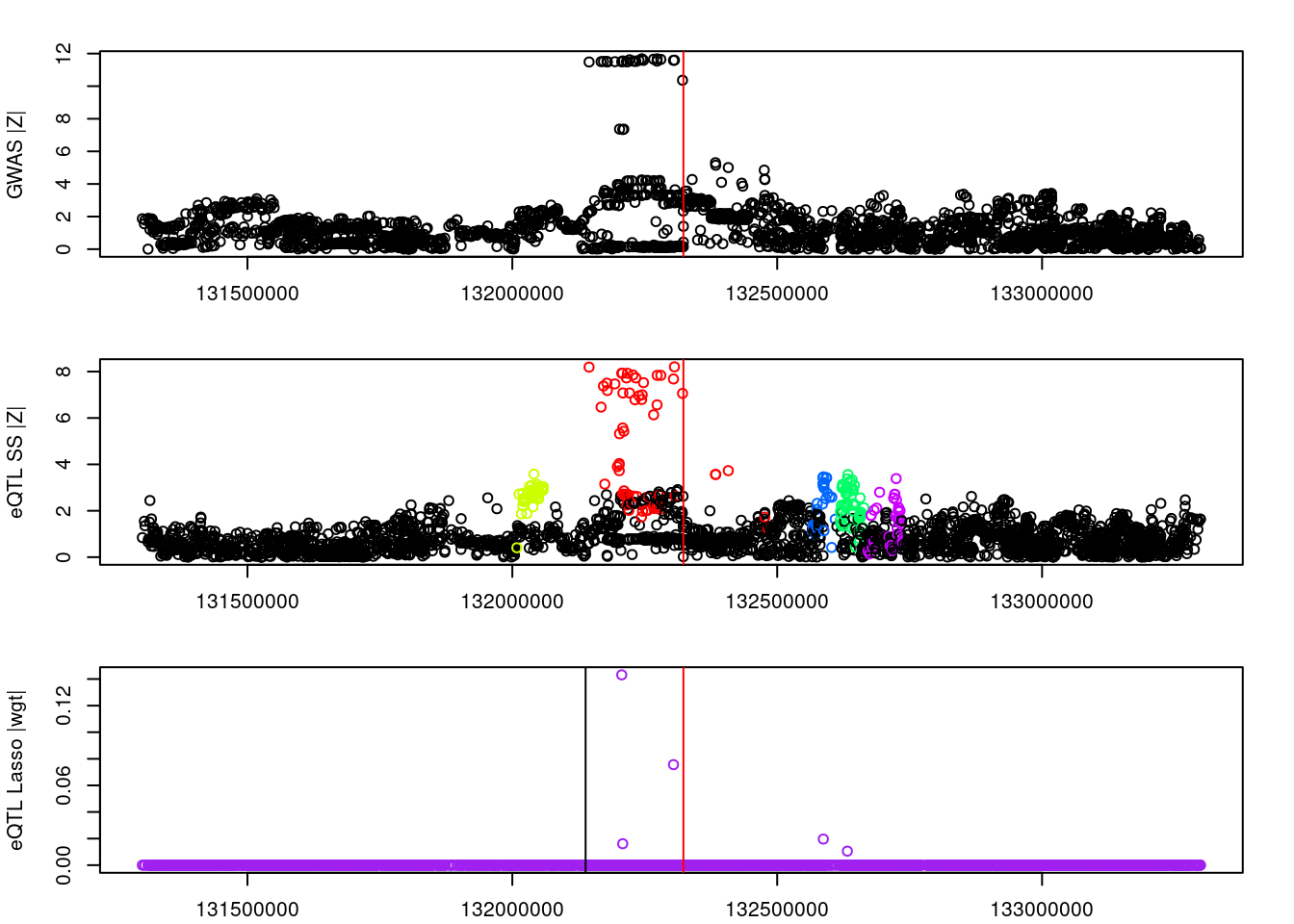
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
4499187 rs11951542 -8.205588 5 132306035 -11.5950440 FALSE 0 1
4499879 rs6893561 3.562638 5 132633568 0.5539217 FALSE 0 3
4498713 rs10069772 3.581654 5 132040688 1.2824300 FALSE 0 2
4499818 rs1560086 3.415952 5 132593410 -1.0714512 FALSE 0 4
4500078 rs11738464 2.460846 5 132725193 2.7154882 FALSE 0 5
trim color
4499187 FALSE #FF0000
4499879 FALSE #00FF66
4498713 TRUE #CCFF00
4499818 TRUE #0066FF
4500078 TRUE #CC00FFpar(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}Warning: There were 2241 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemspar(mfrow=c(1,1))
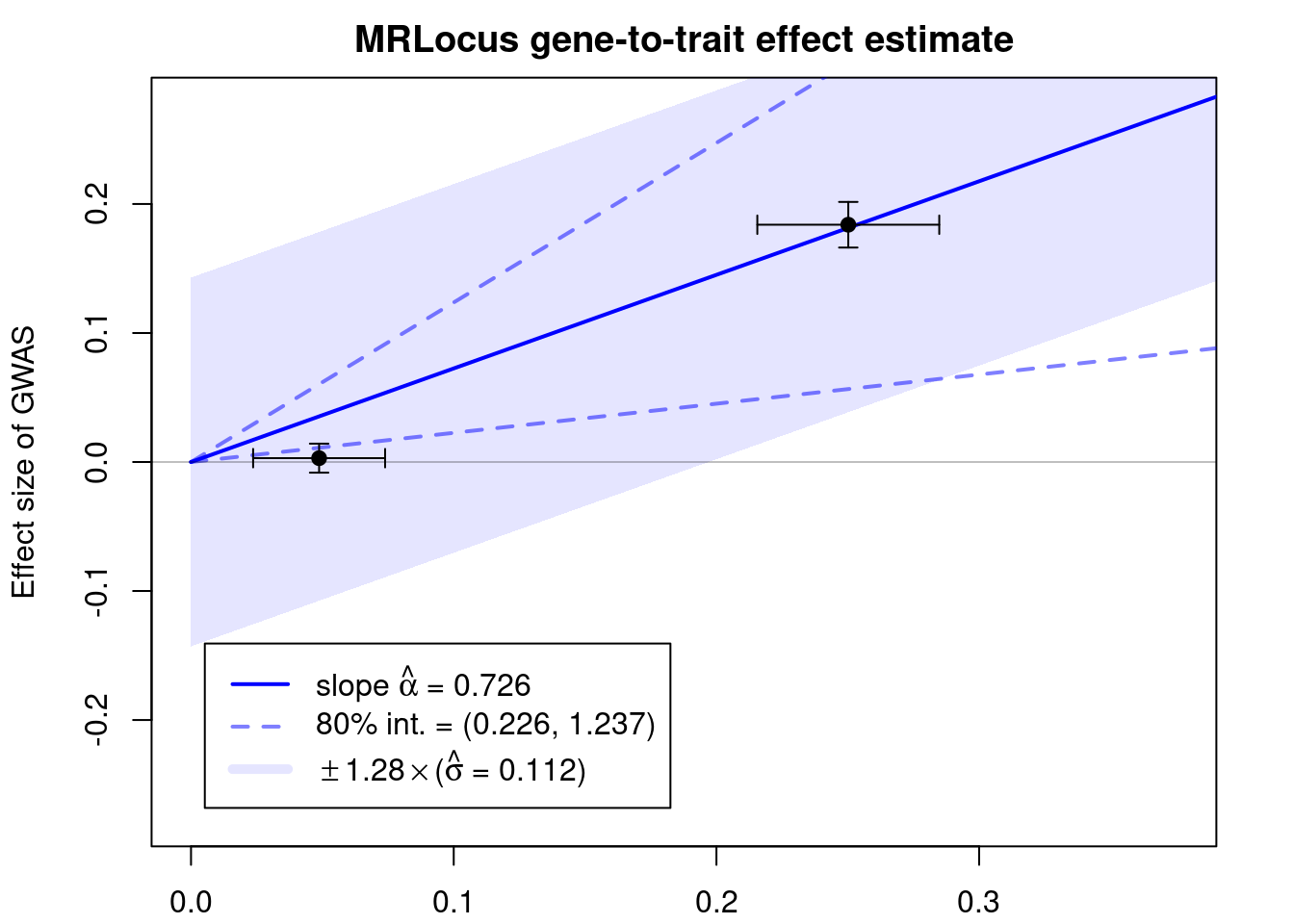
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)Inference for Stan model: slope.
4 chains, each with iter=10000; warmup=5000; thin=1;
post-warmup draws per chain=5000, total post-warmup draws=20000.
mean se_mean sd 10% 90% n_eff Rhat
alpha 0.726 0.005 0.49 0.226 1.237 8293 1
sigma 0.112 0.002 0.10 0.022 0.246 4074 1
Samples were drawn using NUTS(diag_e) at Mon Jun 12 15:26:34 2023.
For each parameter, n_eff is a crude measure of effective sample size,
and Rhat is the potential scale reduction factor on split chains (at
convergence, Rhat=1).if (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
ENSG00000272568
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000272568"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr7_s80.45.2.FUSION7 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000272568.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000272568.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr7_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:27:16 INFO::ENSG00000272568par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)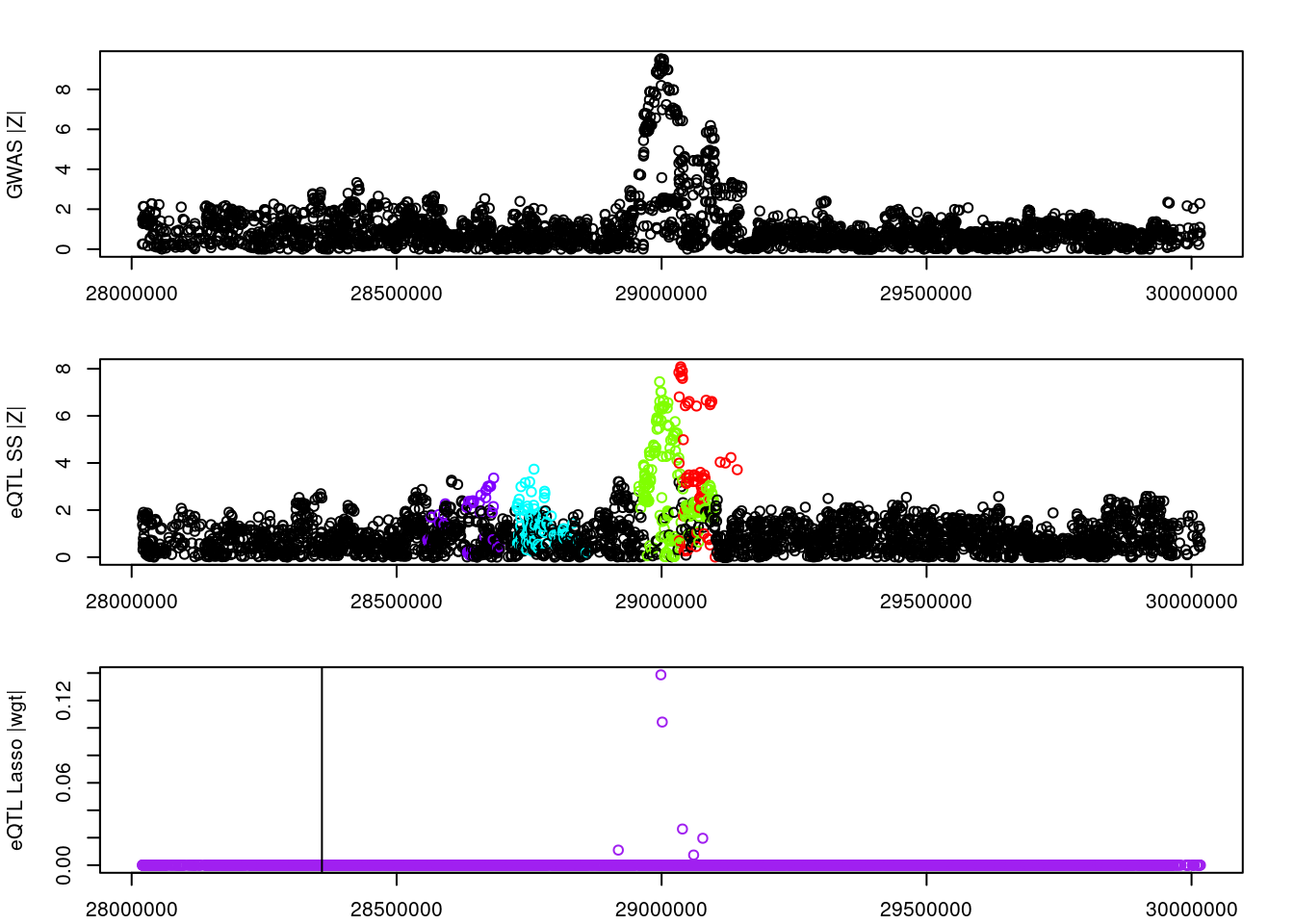
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 71,211,90,48post: 21,50,48,19data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)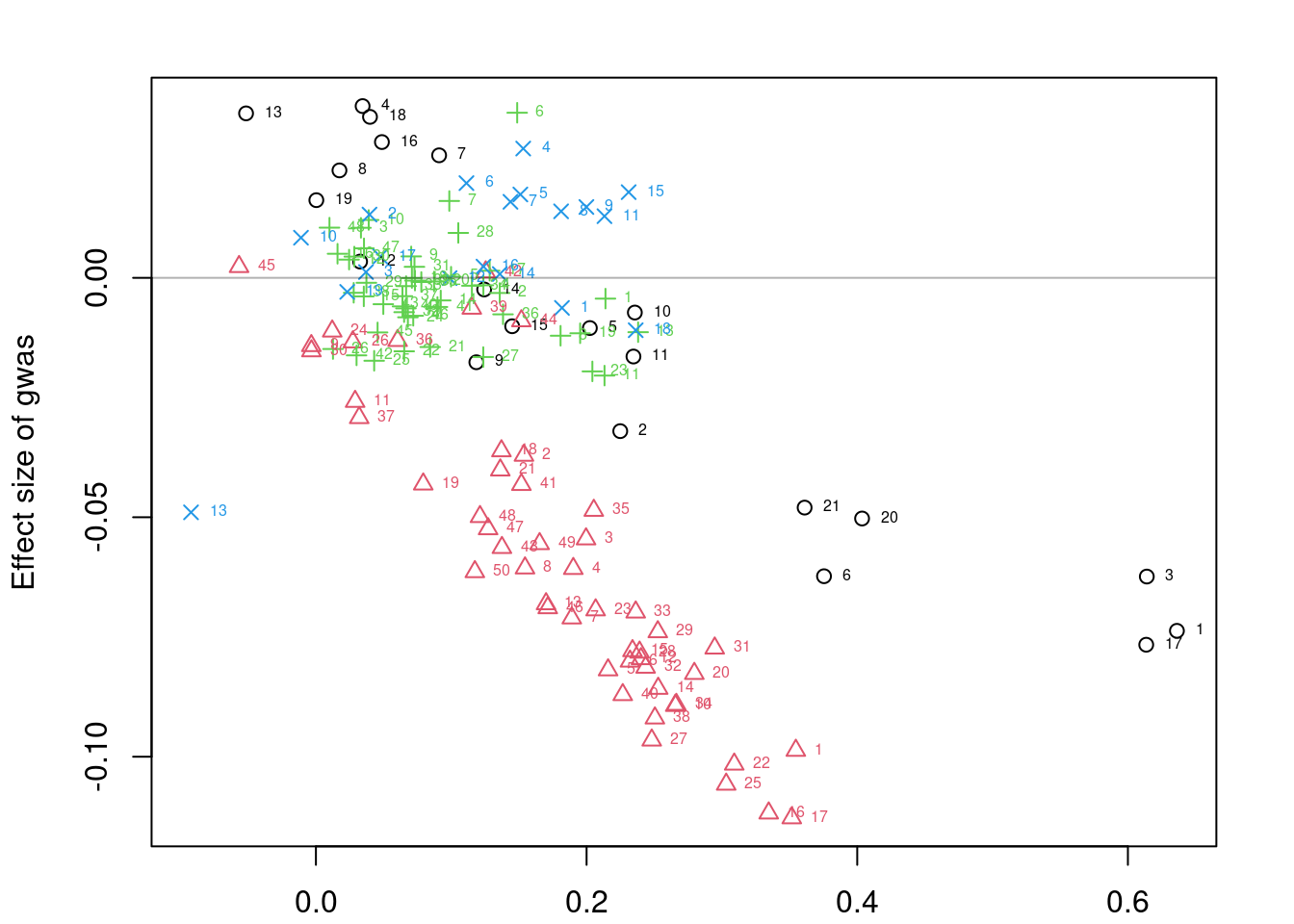
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 538 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 21 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 518 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2805 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 4 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.08, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 862 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2474 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 4 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.09, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 656 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 634 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.07, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)
res$beta_hat_a
[1] 0.42455854 0.29397084 0.06234380 0.04789981
$beta_hat_b
[1] -0.050738650 -0.088684800 -0.001114566 0.001667316
$sd_a
[1] 0.0787349 0.0476176 0.0609834 0.0808790
$sd_b
[1] 0.01665773 0.01042285 0.01254851 0.01625646
$alleles
id ref eff
1 rs11978975 C T
22 rs1006521 G A
90 rs12533079 T G
130 rs73081813 T C#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:30:15 INFO::ENSG00000272568par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)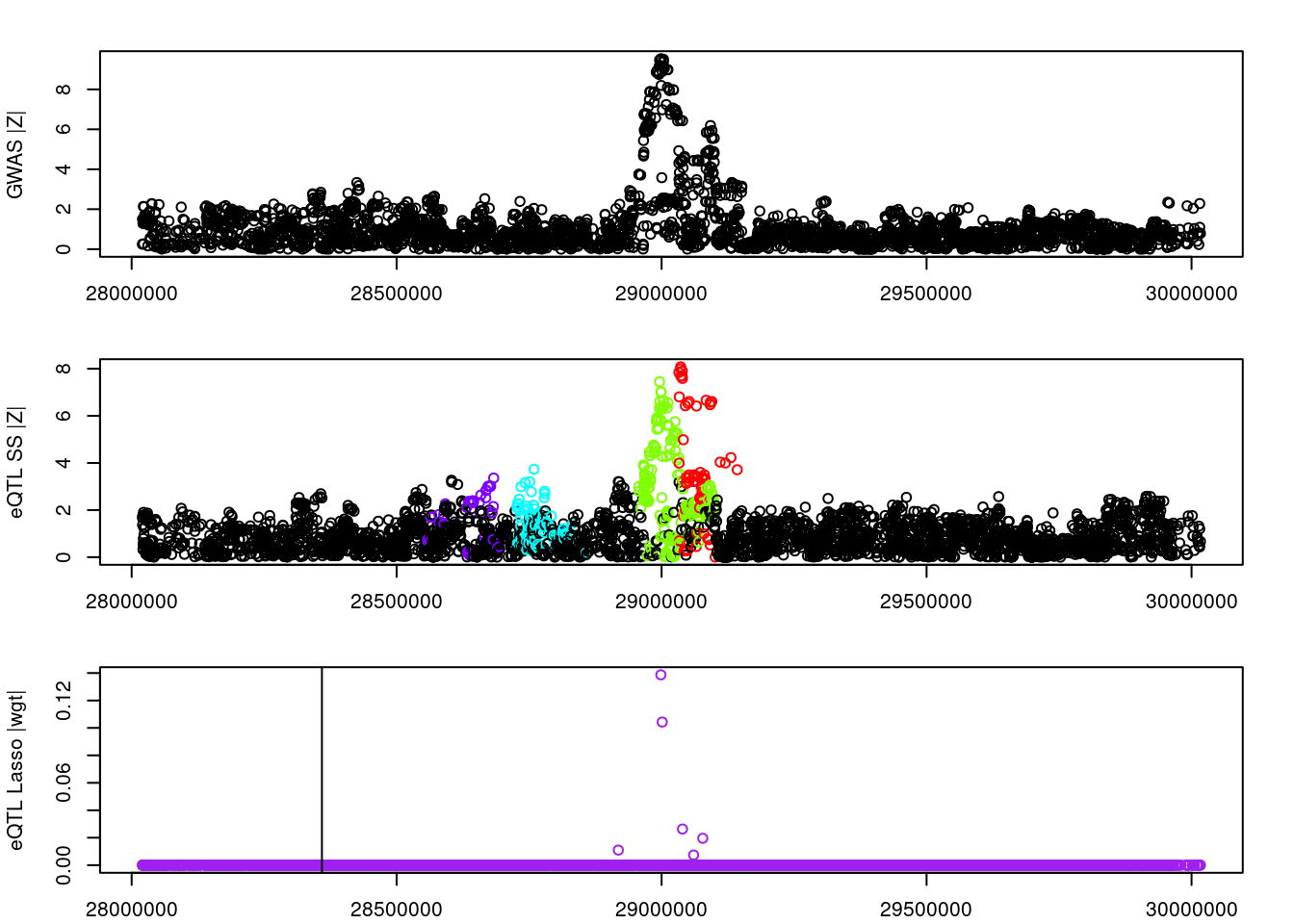
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
5438240 rs1006521 -7.447246 7 28996220 9.4682753 FALSE 0 2
5438362 rs11978975 -8.080584 7 29036108 4.4252724 FALSE 0 1
5437670 rs12533079 -3.201658 7 28751137 0.9224921 FALSE 0 3
5437448 rs73081813 -2.636321 7 28659030 -0.7916247 FALSE 0 4
trim color
5438240 FALSE #80FF00
5438362 TRUE #FF0000
5437670 TRUE #00FFFF
5437448 FALSE #8000FFpar(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}Warning: There were 2317 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-esspar(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)Inference for Stan model: slope.
4 chains, each with iter=10000; warmup=5000; thin=1;
post-warmup draws per chain=5000, total post-warmup draws=20000.
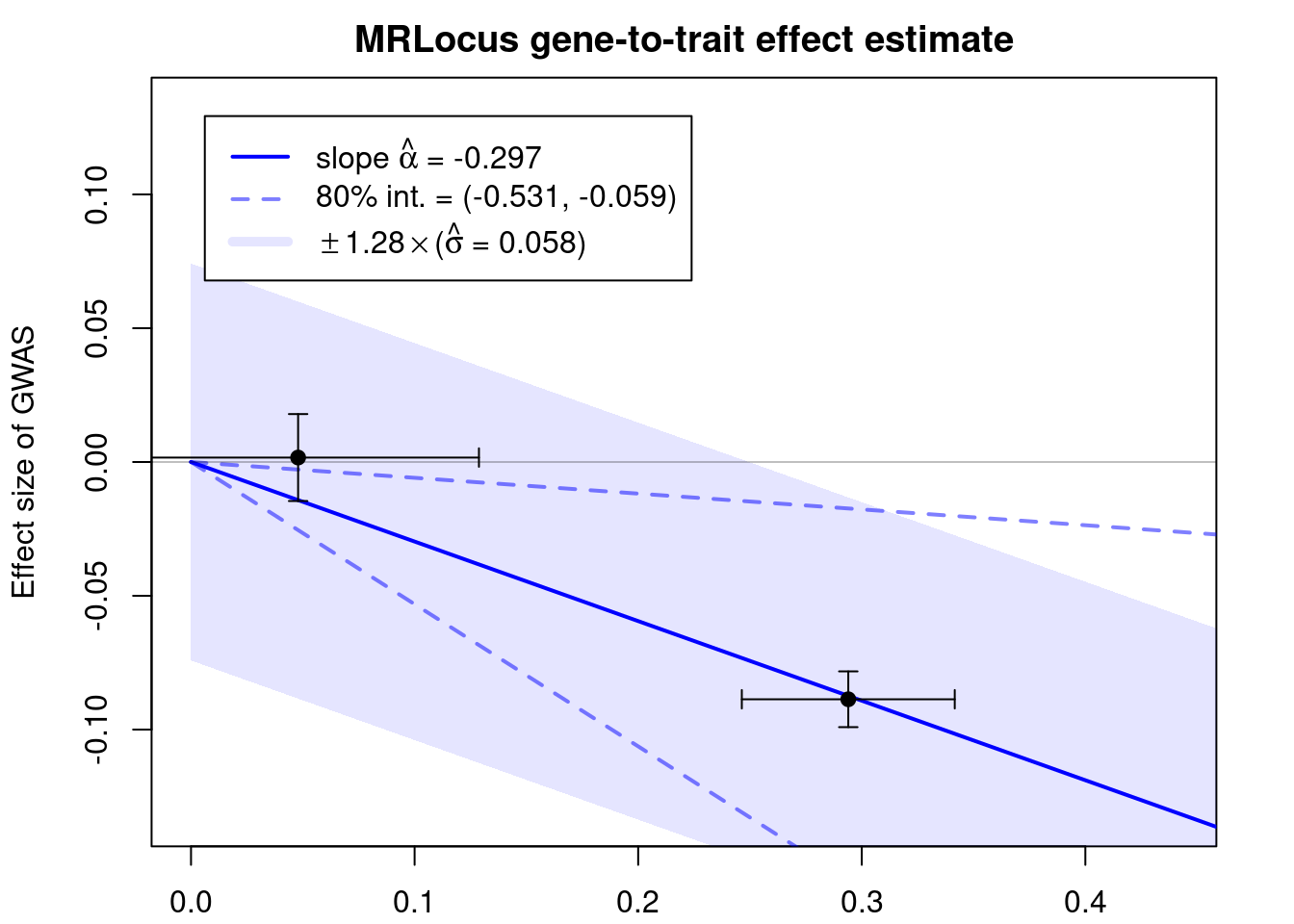
mean se_mean sd 10% 90% n_eff Rhat
alpha -0.297 0.003 0.221 -0.531 -0.059 7275 1.000
sigma 0.058 0.001 0.052 0.009 0.130 1309 1.005
Samples were drawn using NUTS(diag_e) at Mon Jun 12 15:30:17 2023.
For each parameter, n_eff is a crude measure of effective sample size,
and Rhat is the potential scale reduction factor on split chains (at
convergence, Rhat=1).if (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
False positives
ENSG00000243335
set.seed(4099)
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000243335"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr7_s80.45.2.FUSION7 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000243335.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000243335.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr7_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:30:59 INFO::ENSG00000243335par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)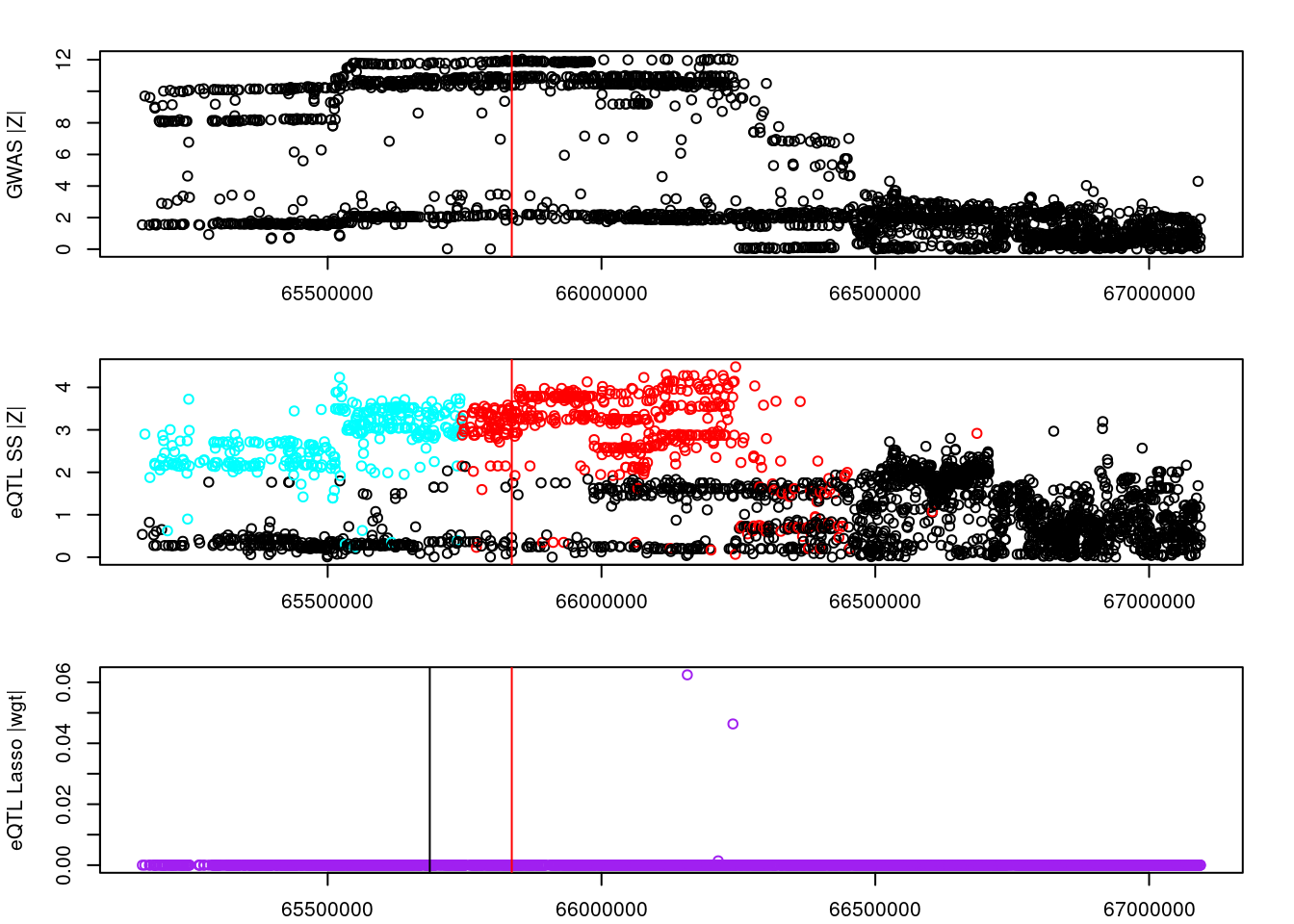
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 541,338post: 37,28data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)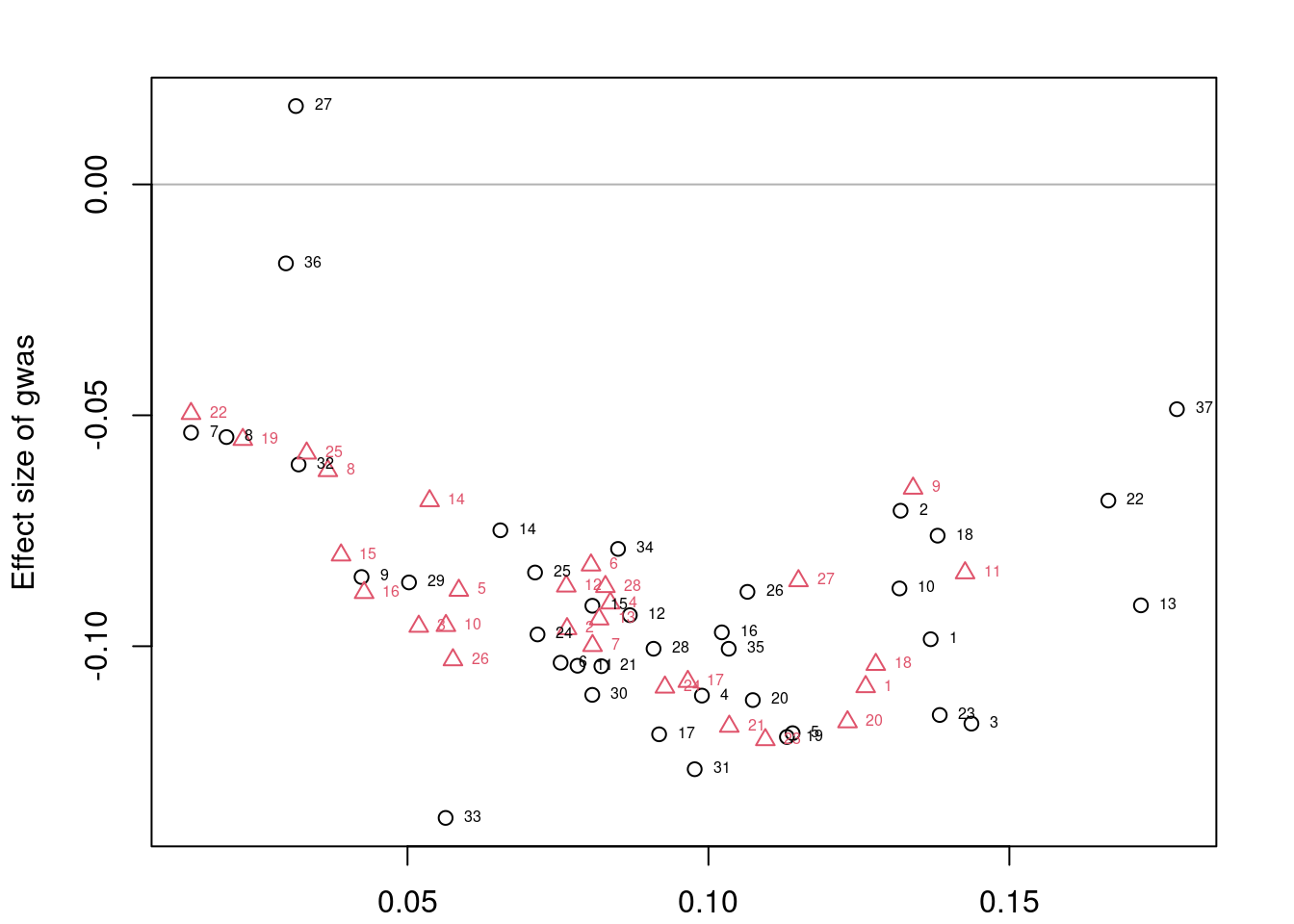
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 775 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 1209 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.06, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 1760 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 49 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.6, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)
res$beta_hat_a
[1] 0.03141847 0.08881357
$beta_hat_b
[1] -0.01188617 -0.06570110
$sd_a
[1] 0.0582605 0.0303630
$sd_b
[1] 0.01968043 0.01070247
$alleles
id ref eff
2 rs116950228 G A
60 rs313829 G A#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:32:10 INFO::ENSG00000243335par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)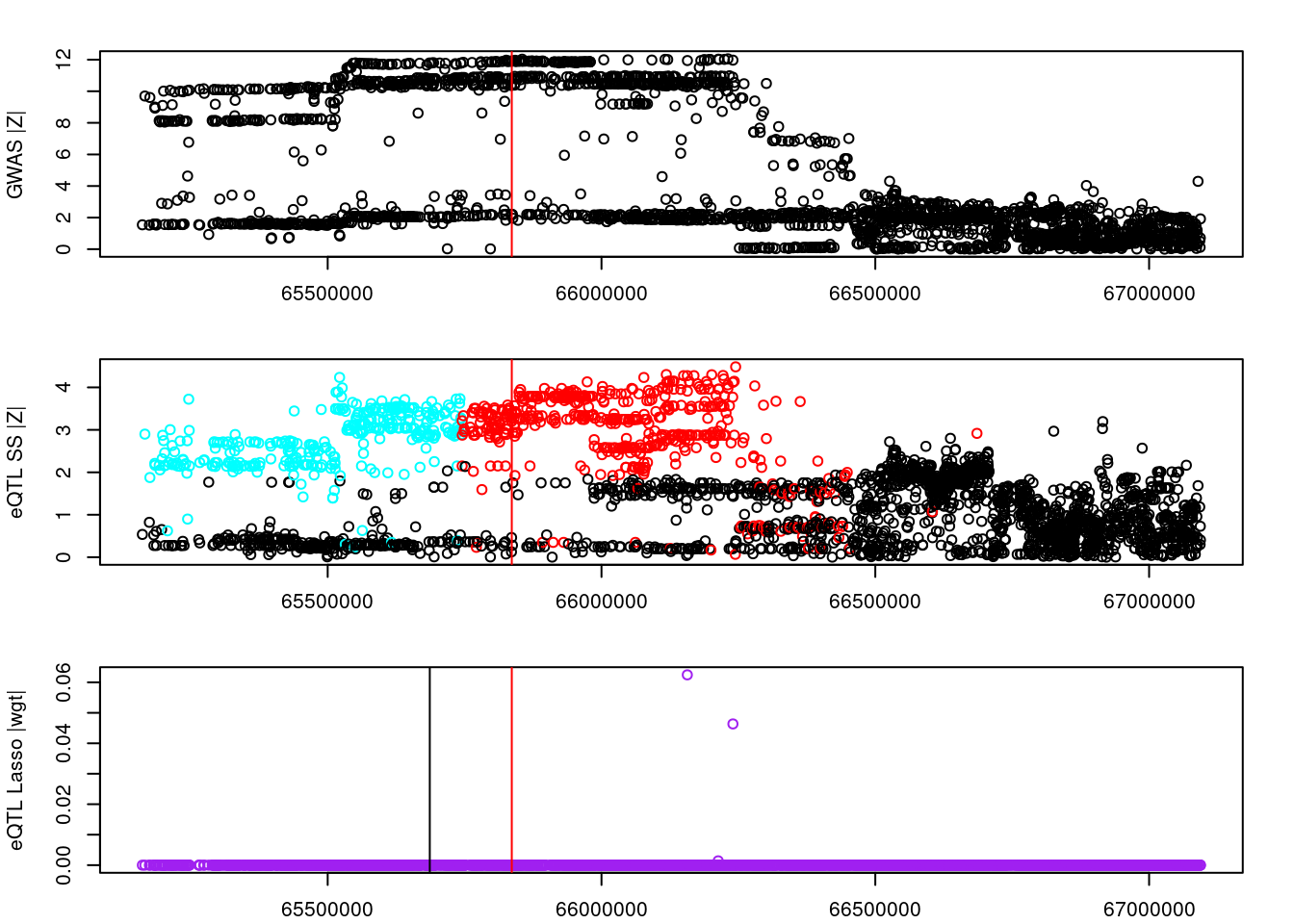
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
5526879 rs313829 3.604848 7 65552497 -11.236654 FALSE 0 2
5528331 rs116950228 -2.264399 7 66327084 3.590381 FALSE 0 1
trim color
5526879 FALSE #00FFFF
5528331 TRUE #FF0000par(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}
par(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)NULLif (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}2023-06-12 15:32:11 INFO::<2 clumps for analysis####################
#investigate LD between confounding variants and eSNPs for clumps
#confounding causal SNPs
df_locus[df_locus$causal,] id z_eqtl chrom pos z_gwas causal weight clump trim
5527360 rs778685 -3.392917 7 65836176 11.90402 TRUE 0 1 FALSE
color
5527360 #FF0000#eSNPs in the analysis
df_locus[match(res$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
5526879 rs313829 3.604848 7 65552497 -11.23665 FALSE 0 2 FALSE
color
5526879 #00FFFF#LD between confounding SNPs and eSNPs
snplist_all <- c(df_locus$id[df_locus$causal], res$alleles$id)
ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
R_snp_info <- lapply(R_snp_info, as.data.frame)
R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
for (j in 1:length(R_snp)){
R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
}
R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
R_snp <- as.matrix(Matrix::bdiag(R_snp))
colnames(R_snp) <- R_snp_info$id
rownames(R_snp) <- R_snp_info$id
#number of LD regions
length(ld_R_idx)[1] 2#LD between variants
R_snp[snplist_all, snplist_all] rs778685 rs313829
rs778685 1 0
rs313829 0 1False negatives
ENSG00000215022
set.seed(45102)
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000215022"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr6_s80.45.2.FUSION6 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000215022.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000215022.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr6_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:32:58 INFO::ENSG00000215022par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)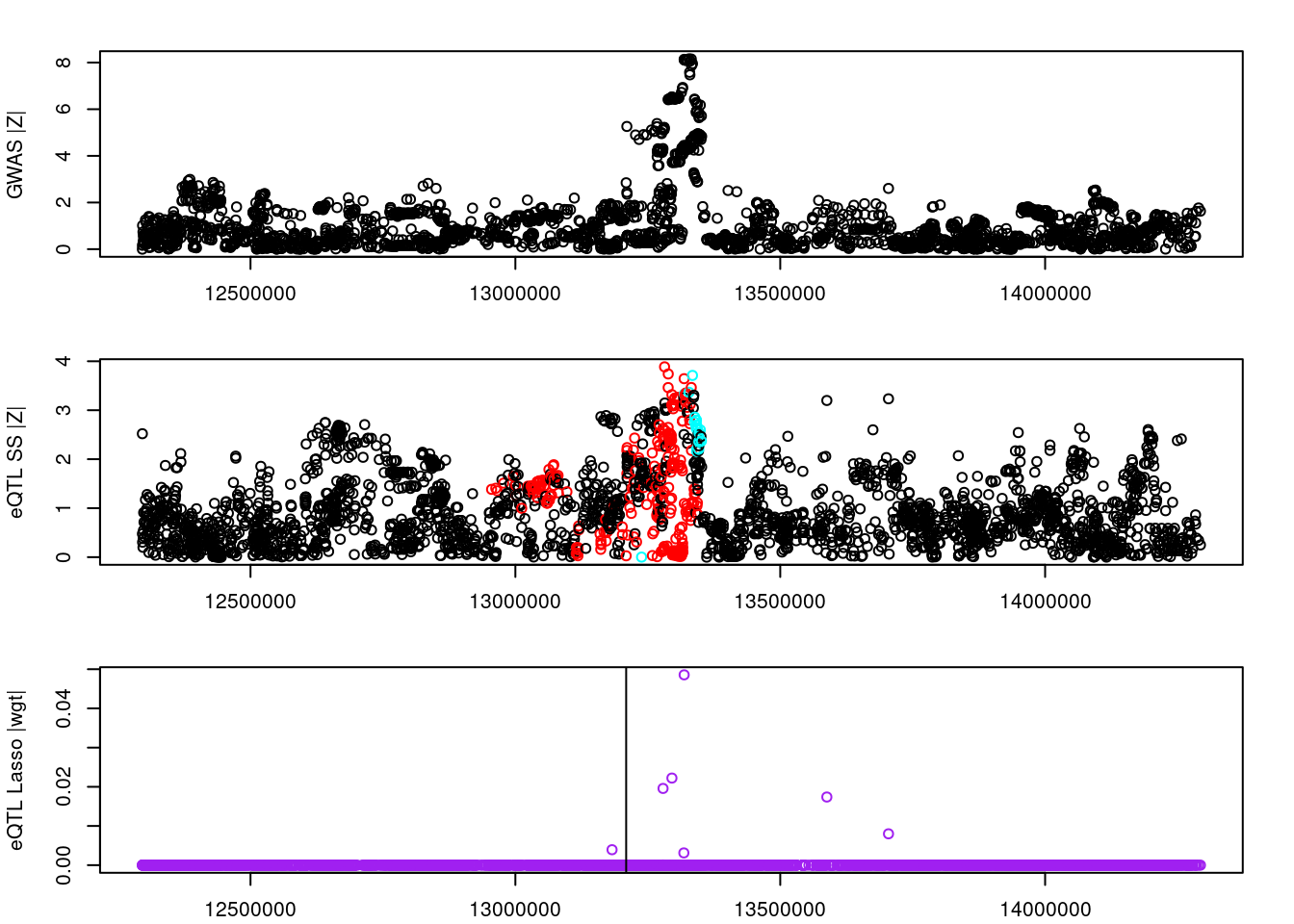
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 312,21post: 45,04data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)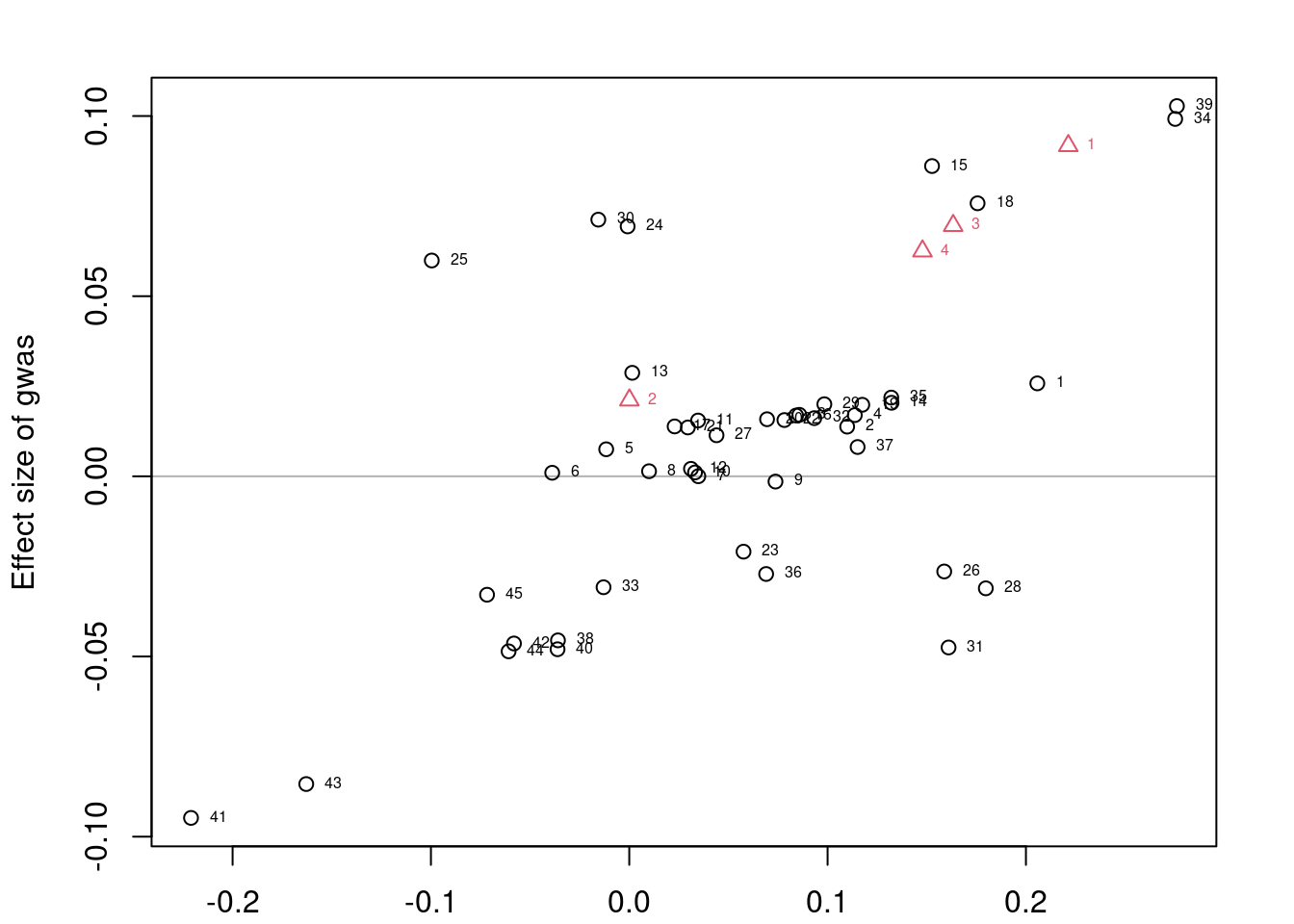
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 1355 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 1086 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.28, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 348 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.1, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)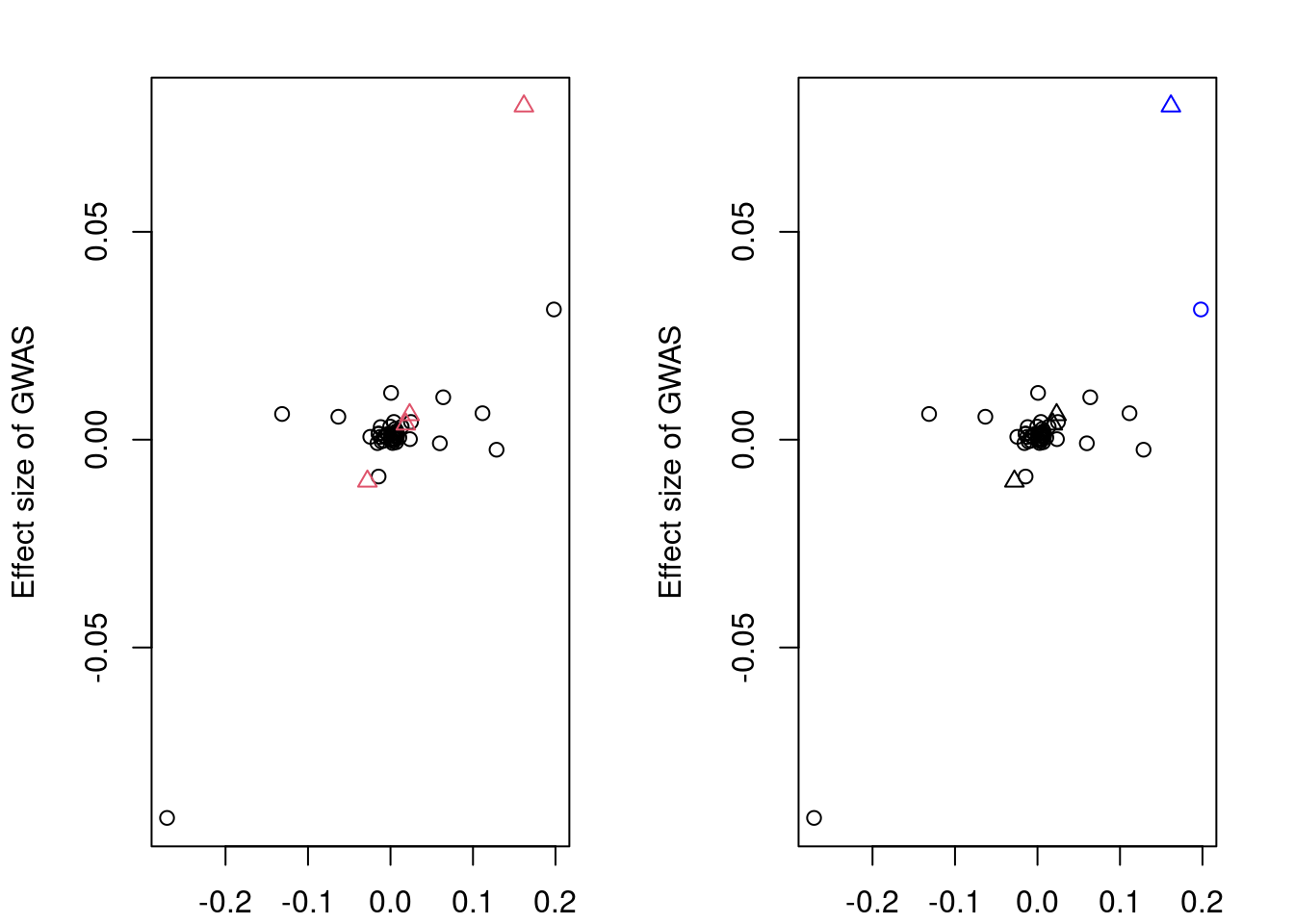
res$beta_hat_a
[1] 0.1980562 0.1617230
$beta_hat_b
[1] 0.03131939 0.08024581
$sd_a
[1] 0.0846188 0.0596666
$sd_b
[1] 0.01583697 0.01153973
$alleles
id ref eff
39 rs3800484 T C
46 rs2439556 A G#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:34:01 INFO::ENSG00000215022par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)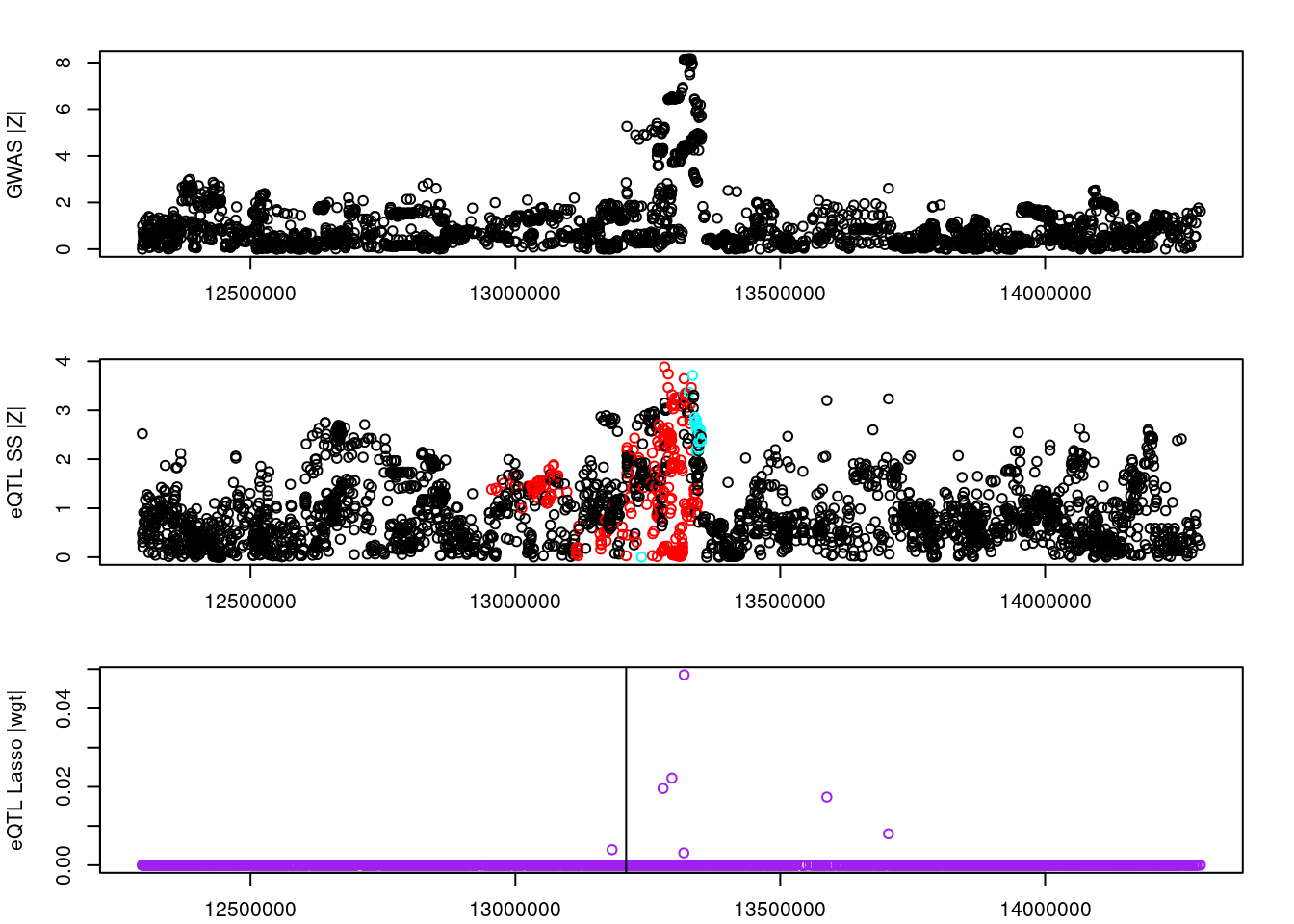
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
4897517 rs2439556 -3.709932 6 13334307 -7.952074 FALSE 0 2 FALSE
4897409 rs3800484 3.263211 6 13307046 6.488619 FALSE 0 1 TRUE
color
4897517 #00FFFF
4897409 #FF0000par(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}
par(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)NULLif (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}2023-06-12 15:34:02 INFO::<2 clumps for analysis####################
#investigate LD between variants with largest weights and eSNPs for clumps
#variants with largest weight
df_locus <- df_locus[order(-abs(df_locus$weight)),]
df_locus[df_locus$weight!=0,] id z_eqtl chrom pos z_gwas causal weight clump
4897476 rs555100 -3.643406 6 13318609 -8.138288 FALSE 4.85653e-02 1
4897349 rs1550526 -3.317257 6 13295515 -6.491346 FALSE -2.22233e-02 1
4897261 rs9395602 2.626244 6 13278753 -2.367597 FALSE -1.95757e-02 1
4897980 rs2841512 -3.198471 6 13587980 -1.908055 FALSE -1.73881e-02 0
4898103 rs204221 -3.232695 6 13704341 -2.604934 FALSE -7.99743e-03 0
4868316 rs1150609 -2.721145 6 13182501 -1.841752 FALSE 3.94201e-03 0
4897473 rs3823441 3.256555 6 13318030 4.418932 FALSE -3.13081e-03 0
4897262 rs9381884 2.626244 6 13278788 -2.361500 FALSE -5.09785e-05 1
trim color
4897476 FALSE #FF0000
4897349 FALSE #FF0000
4897261 FALSE #FF0000
4897980 FALSE black
4898103 FALSE black
4868316 FALSE black
4897473 FALSE black
4897262 FALSE #FF0000#eSNPs in the analysis
df_locus[match(res$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
4897517 rs2439556 -3.709932 6 13334307 -7.952074 FALSE 0 2 FALSE
color
4897517 #00FFFF#LD between confounding SNPs and eSNPs
snplist_all <- c(df_locus$id[df_locus$weight!=0], res$alleles$id)
ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
R_snp_info <- lapply(R_snp_info, as.data.frame)
R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
for (j in 1:length(R_snp)){
R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
}
R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
R_snp <- as.matrix(Matrix::bdiag(R_snp))
colnames(R_snp) <- R_snp_info$id
rownames(R_snp) <- R_snp_info$id
#number of LD regions
length(ld_R_idx)[1] 2#LD between variants
R_snp[snplist_all, snplist_all] rs555100 rs1550526 rs9395602 rs2841512 rs204221
rs555100 1.00000000 0.18517096 0.505815561 0.023015009 0.04337648
rs1550526 0.18517096 1.00000000 0.168641522 -0.026414426 0.02045739
rs9395602 0.50581556 0.16864152 1.000000000 -0.008500891 -0.02706590
rs2841512 0.02301501 -0.02641443 -0.008500891 1.000000000 0.77255548
rs204221 0.04337648 0.02045739 -0.027065899 0.772555478 1.00000000
rs1150609 0.00000000 0.00000000 0.000000000 0.000000000 0.00000000
rs3823441 -0.18818972 -0.47797099 -0.029256809 0.049136194 0.01666876
rs9381884 0.50581556 0.16864152 1.000000000 -0.008500891 -0.02706590
rs2439556 0.98146958 0.18700369 0.496965223 0.029135961 0.04911024
rs1150609 rs3823441 rs9381884 rs2439556
rs555100 0 -0.18818972 0.505815561 0.98146958
rs1550526 0 -0.47797099 0.168641522 0.18700369
rs9395602 0 -0.02925681 1.000000000 0.49696522
rs2841512 0 0.04913619 -0.008500891 0.02913596
rs204221 0 0.01666876 -0.027065899 0.04911024
rs1150609 1 0.00000000 0.000000000 0.00000000
rs3823441 0 1.00000000 -0.029256809 -0.18683534
rs9381884 0 -0.02925681 1.000000000 0.49696522
rs2439556 0 -0.18683534 0.496965223 1.00000000ENSG00000184898
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000184898"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr2_s80.45.2.FUSION2 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000184898.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000184898.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr2_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:34:44 INFO::ENSG00000184898par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)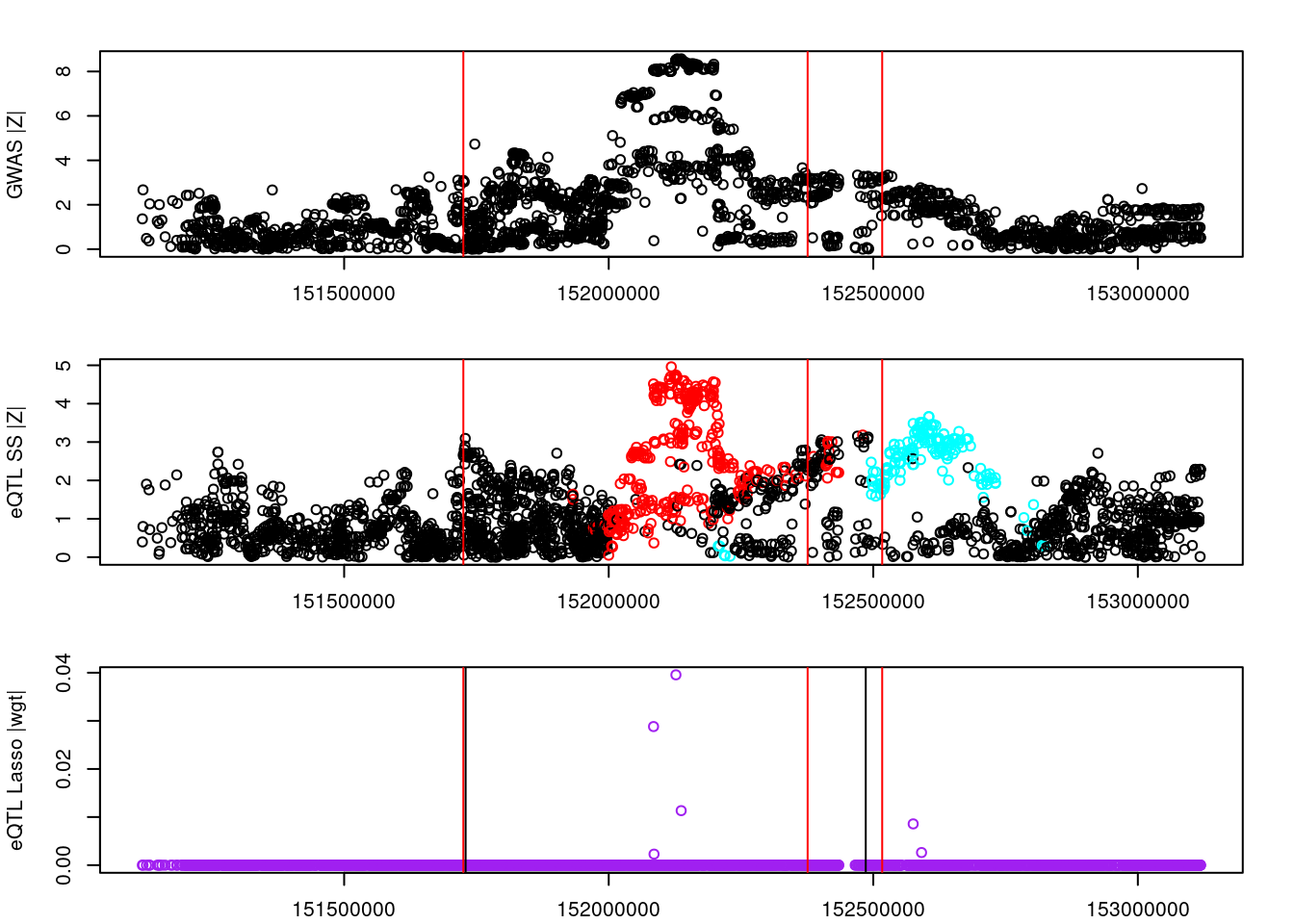
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 396,164post: 59,19data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 1013 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2307 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 4 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.23, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 1254 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 95 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.36, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)
res$beta_hat_a
[1] 0.08661205 0.02370354
$beta_hat_b
[1] -0.0349405113 0.0006716198
$sd_a
[1] 0.0282915 0.0413816
$sd_b
[1] 0.009899139 0.017600476
$alleles
id ref eff
31 rs11730 G A
60 rs9677576 A G#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:36:29 INFO::ENSG00000184898par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)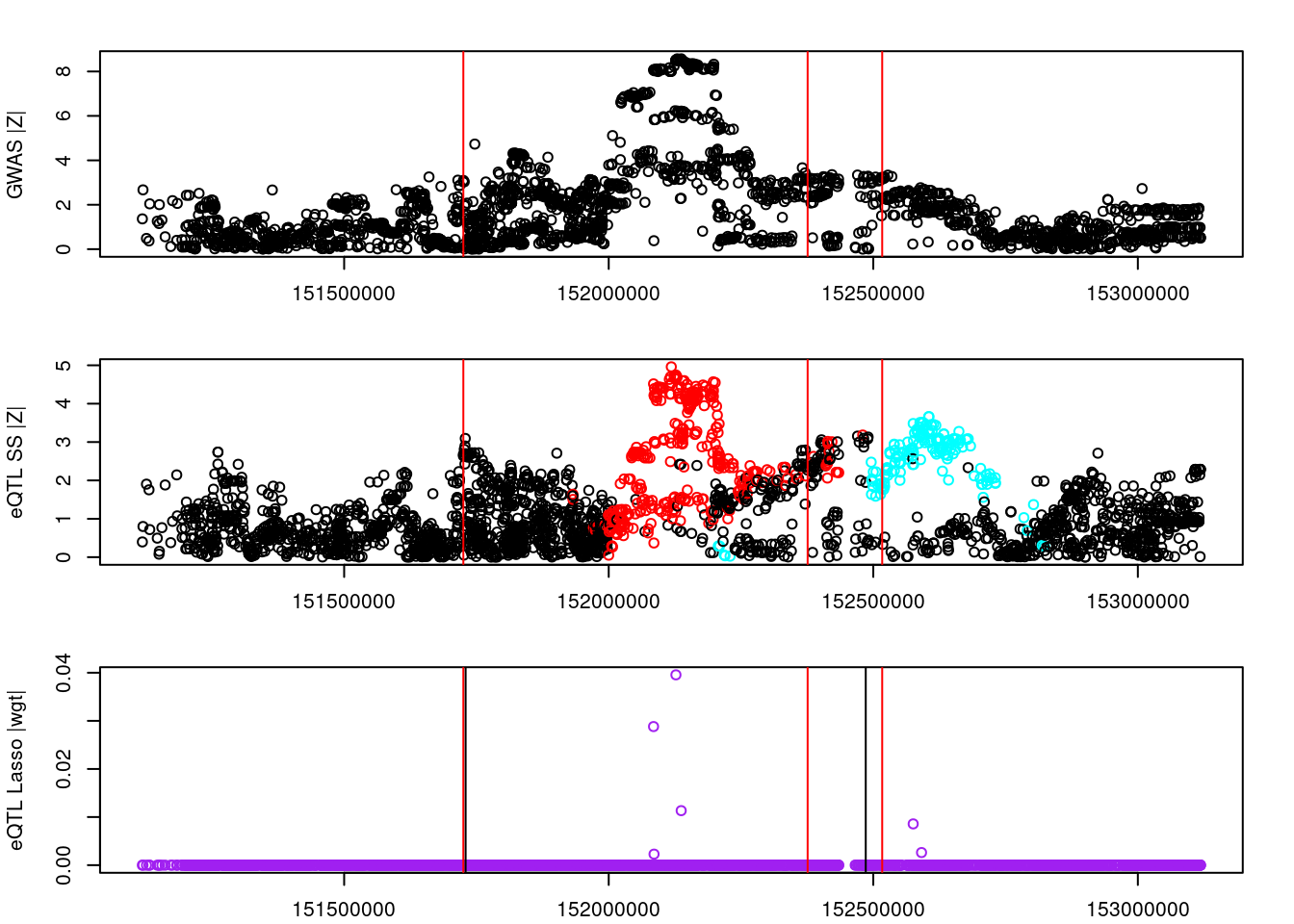
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
2813724 rs11730 4.745171 2 152127011 -8.513674 FALSE -0.0395602 1
2814517 rs9677576 3.661217 2 152604697 1.890673 FALSE 0.0000000 2
trim color
2813724 FALSE #FF0000
2814517 TRUE #00FFFFpar(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}
par(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)NULLif (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}2023-06-12 15:36:30 INFO::<2 clumps for analysis####################
#investigate LD between variants with largest weights and eSNPs for clumps
#variants with largest weight
df_locus <- df_locus[order(-abs(df_locus$weight)),]
df_locus[df_locus$weight!=0,] id z_eqtl chrom pos z_gwas causal weight
2813724 rs11730 4.745171 2 152127011 -8.513674 FALSE -3.95602e-02
2813649 rs289903 -4.515707 2 152084705 8.062208 FALSE -2.88088e-02
2813751 rs3856557 4.435029 2 152136961 -8.485281 FALSE -1.13303e-02
2814465 rs4664498 3.482618 2 152575417 1.839083 FALSE 8.57801e-03
2814492 rs11896217 3.508597 2 152591189 2.124467 FALSE 2.62155e-03
2813651 rs289904 -4.224670 2 152085643 8.087215 FALSE -2.29195e-03
2814497 rs4393745 3.508597 2 152594206 2.154522 FALSE 6.82696e-06
clump trim color
2813724 1 FALSE #FF0000
2813649 1 FALSE #FF0000
2813751 1 FALSE #FF0000
2814465 2 FALSE #00FFFF
2814492 2 FALSE #00FFFF
2813651 1 FALSE #FF0000
2814497 2 FALSE #00FFFF#eSNPs in the analysis
df_locus[match(res$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
2813724 rs11730 4.745171 2 152127011 -8.513674 FALSE -0.0395602 1
trim color
2813724 FALSE #FF0000#LD between confounding SNPs and eSNPs
snplist_all <- c(df_locus$id[df_locus$weight!=0], res$alleles$id)
ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
R_snp_info <- lapply(R_snp_info, as.data.frame)
R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
for (j in 1:length(R_snp)){
R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
}
R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
R_snp <- as.matrix(Matrix::bdiag(R_snp))
colnames(R_snp) <- R_snp_info$id
rownames(R_snp) <- R_snp_info$id
#number of LD regions
length(ld_R_idx)[1] 2#LD between variants
R_snp[snplist_all, snplist_all] rs11730 rs289903 rs3856557 rs4664498 rs11896217 rs289904
rs11730 1.0000000 -0.9055700 0.9992733 0.0000000 0.0000000 -0.9050076
rs289903 -0.9055700 1.0000000 -0.9042423 0.0000000 0.0000000 0.9988528
rs3856557 0.9992733 -0.9042423 1.0000000 0.0000000 0.0000000 -0.9036801
rs4664498 0.0000000 0.0000000 0.0000000 1.0000000 0.8924160 0.0000000
rs11896217 0.0000000 0.0000000 0.0000000 0.8924160 1.0000000 0.0000000
rs289904 -0.9050076 0.9988528 -0.9036801 0.0000000 0.0000000 1.0000000
rs4393745 0.0000000 0.0000000 0.0000000 0.8919337 0.9999043 0.0000000
rs11730 1.0000000 -0.9055700 0.9992733 0.0000000 0.0000000 -0.9050076
rs4393745 rs11730
rs11730 0.0000000 1.0000000
rs289903 0.0000000 -0.9055700
rs3856557 0.0000000 0.9992733
rs4664498 0.8919337 0.0000000
rs11896217 0.9999043 0.0000000
rs289904 0.0000000 -0.9050076
rs4393745 1.0000000 0.0000000
rs11730 0.0000000 1.0000000False positives
ENSG00000243335
set.seed(13450)
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000243335"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr7_s80.45.2.FUSION7 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000243335.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000243335.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr7_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:37:13 INFO::ENSG00000243335par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)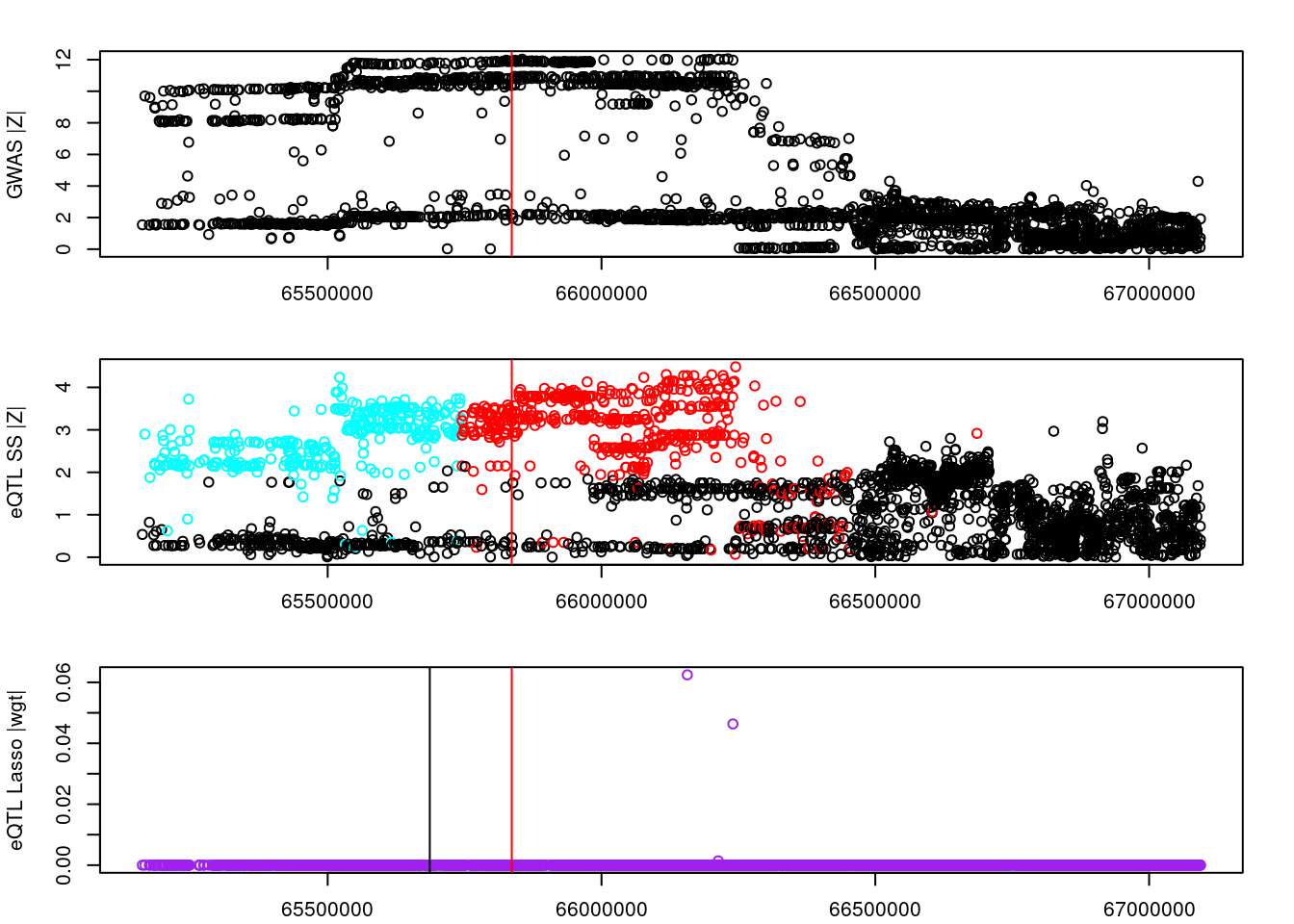
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 541,338post: 37,28data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 1153 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 225 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.2, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 658 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 236 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 3 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
res$beta_hat_a
[1] 0.02560447 0.05262674
$beta_hat_b
[1] -0.02139649 -0.04498408
$sd_a
[1] 0.0339436 0.0303630
$sd_b
[1] 0.01124993 0.01070247
$alleles
id ref eff
3 rs28817617 G A
60 rs313829 G A#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:38:13 INFO::ENSG00000243335par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)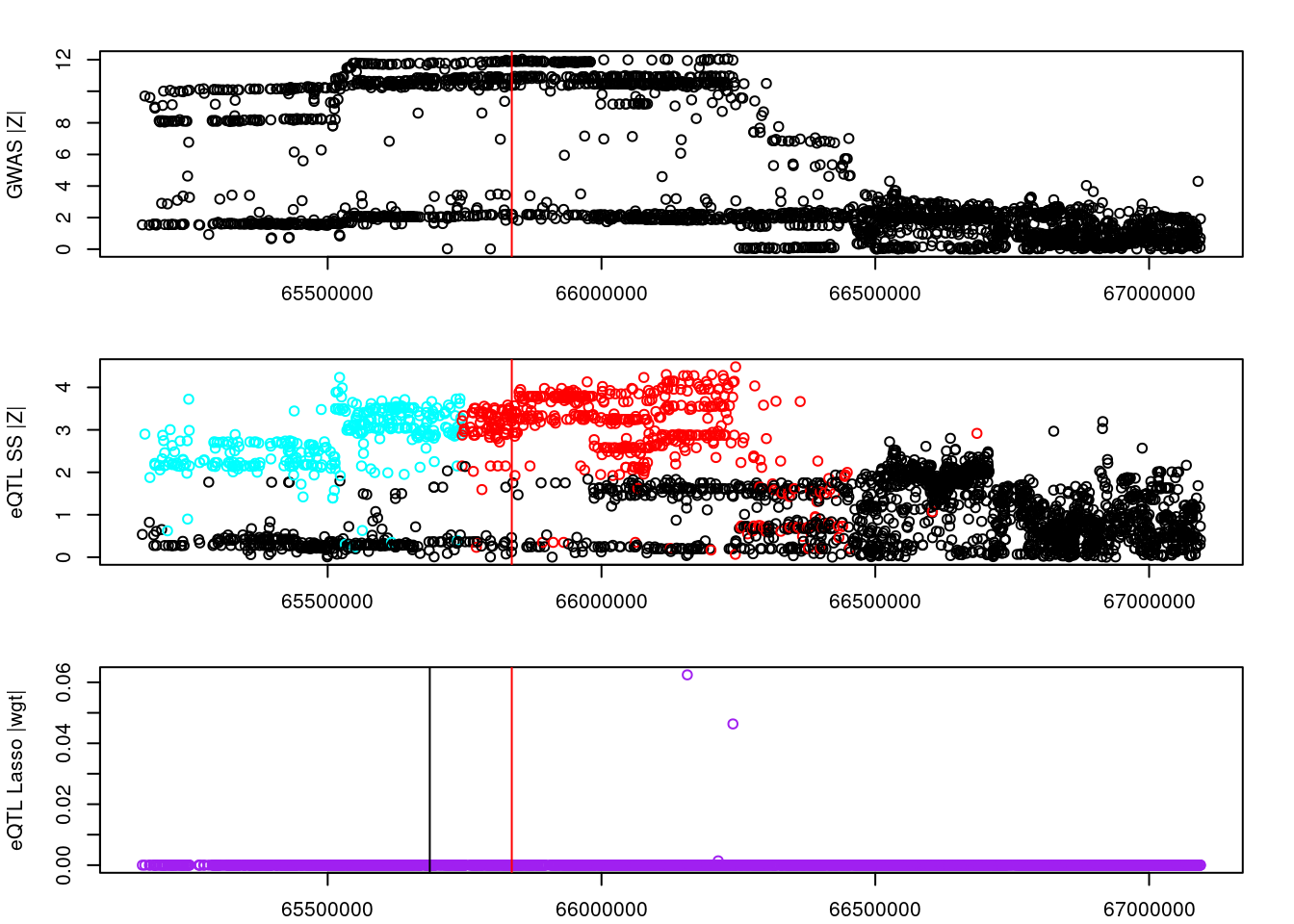
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
5526879 rs313829 3.604848 7 65552497 -11.23665 FALSE 0 2 FALSE
5527815 rs28817617 4.233434 7 66077355 -10.37869 FALSE 0 1 TRUE
color
5526879 #00FFFF
5527815 #FF0000par(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}
par(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)NULLif (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}2023-06-12 15:38:14 INFO::<2 clumps for analysis####################
#investigate LD between confounding variants and eSNPs for clumps
#confounding causal SNPs
df_locus[df_locus$causal,] id z_eqtl chrom pos z_gwas causal weight clump trim
5527360 rs778685 -3.392917 7 65836176 11.90402 TRUE 0 1 FALSE
color
5527360 #FF0000#eSNPs in the analysis
df_locus[match(res$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
5526879 rs313829 3.604848 7 65552497 -11.23665 FALSE 0 2 FALSE
color
5526879 #00FFFF#LD between confounding SNPs and eSNPs
snplist_all <- c(df_locus$id[df_locus$causal], res$alleles$id)
ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
R_snp_info <- lapply(R_snp_info, as.data.frame)
R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
for (j in 1:length(R_snp)){
R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
}
R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
R_snp <- as.matrix(Matrix::bdiag(R_snp))
colnames(R_snp) <- R_snp_info$id
rownames(R_snp) <- R_snp_info$id
#number of LD regions
length(ld_R_idx)[1] 2#LD between variants
R_snp[snplist_all, snplist_all] rs778685 rs313829
rs778685 1 0
rs313829 0 1ENSG00000237310
load("/home/wcrouse/scratch-midway2/mrlocus_image.RData")
####################
#run MR-Locus
gene <- "ENSG00000237310"
chr <- as.numeric(gtf_df$seqnames[gtf_df$gene_id==gene])
#prepare files for plink
plink_exclude_file <- paste0(temp_dir, "ukb_chr", chr, "_s80.45.2.exclude")
if (!file.exists(plink_exclude_file)){
#store list of SNPs in .bed file using plink
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --write-snplist --out ",
temp_dir, "ukb_chr", chr, "_s80.45.2")
system(system_cmd)
#store list of duplicate SNPs in .bed file to exclude
system_cmd <- paste0("sort ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.snplist|uniq -d > ",
temp_dir,
"ukb_chr", chr, "_s80.45.2.exclude")
system(system_cmd)
}
#clumping using plink
eqtl_current <- eqtl[eqtl$gene_id==gene,]
eqtl_current <- dplyr::rename(eqtl_current, SNP="id", P="pval_nominal")
eqtl_current_file <- paste0(temp_dir, outname_base, ".eqtl_sumstats.", gene, ".temp")
write.table(eqtl_current, file=eqtl_current_file, sep="\t", col.names=T, row.names=F, quote=F)
eqtl_clump_file <- paste0(temp_dir, outname_base, ".eqtl_clumps.", gene, ".temp")
system_cmd <- paste0("plink --bfile ",
ld_bedfs$file[chr],
" --clump ",
eqtl_current_file,
" --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out ",
eqtl_clump_file,
" --exclude ",
plink_exclude_file)
#system(system_cmd)
print(system_cmd)[1] "plink --bfile /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/ukb_chr7_s80.45.2.FUSION7 --clump /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_sumstats.ENSG00000237310.temp --clump-p1 0.001 --clump-p2 1 --clump-r2 0.1 --clump-kb 500 --out /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/mrlocus_report_temp.eqtl_clumps.ENSG00000237310.temp --exclude /home/wcrouse/causalTWAS/simulations/simulation_ctwas_rss_20210416_compare/temp_mrlocus/ukb_chr7_s80.45.2.exclude"eqtl_clump_file <- paste0(eqtl_clump_file, ".clumped")
clumps <- read.table(eqtl_clump_file, header=T)
clumps$SP2 <- sapply(1:nrow(clumps), function(x){paste0(clumps$SNP[x], "(1),", clumps$SP2[x])})
clumps <- lapply(clumps$SP2, function(y){unname(sapply(unlist(strsplit(y, ",")), function(x){unlist(strsplit(x, "[(]"))[1]}))})
clumps <- lapply(clumps, function(x){x[x!="NONE"]})
####################
df_locus <- data.frame(id=eqtl_current$SNP, z_eqtl=eqtl_current$slope/eqtl_current$slope_se)
df_locus <- cbind(df_locus, ld_R_info[match(df_locus$id, ld_R_info$id),c("chrom","pos")])
df_locus$z_gwas <- (gwas$Estimate/gwas$Std.Error)[match(df_locus$id, gwas$id)]
df_locus$causal <- df_locus$id %in% names(true_snp_effects)
weightf_dir <- "/project2/mstephens/causalTWAS/fusion_weights/Adipose_Subcutaneous/"
weightf_gene <- list.files(weightf_dir)
weightf_gene <- paste0(weightf_dir, weightf_gene[grep(gene, weightf_gene)])
load(weightf_gene)
df_locus$weight <- wgt.matrix[match(df_locus$id, rownames(wgt.matrix)),"lasso"]
df_locus$weight[is.na(df_locus$weight)] <- 0
df_locus$clump <- 0
for (j in 1:length(clumps)){
df_locus$clump[df_locus$id %in% clumps[[j]]] <- j
}
#visualize input data
colors <- c("black", rainbow(length(clumps)))
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:39:02 INFO::ENSG00000237310par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)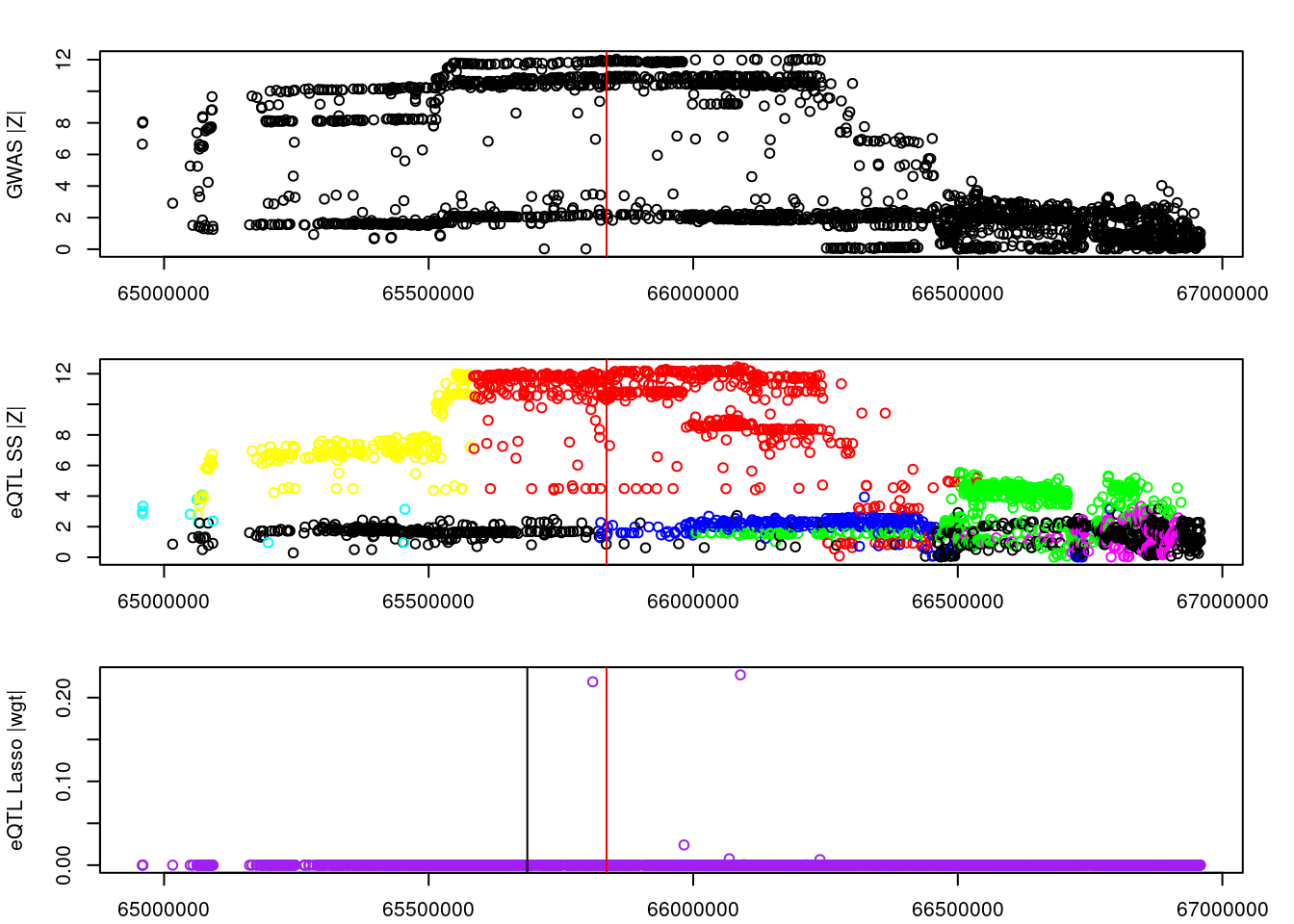
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
# #load LD matrix for SNPs in all clumps
# snplist_all <- unlist(clumps)
#
# ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
#
# R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
#
# R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
# R_snp_info <- lapply(R_snp_info, as.data.frame)
#
# R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
# rm(snplist_all)
#
# for (j in 1:length(R_snp)){
# R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
# R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
# }
#
# R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
# R_snp <- as.matrix(Matrix::bdiag(R_snp))
#
# colnames(R_snp) <- R_snp_info$id
# rownames(R_snp) <- R_snp_info$id
#
#
#
# R_snp_bkup <- R_snp
####################
#load genotypes and compute LD matrix for SNPs in all clumps
snplist_all <- unlist(clumps)
ld_pvarf <- ld_pvarfs[chr]
ldref_file <- ldref_files[chr]
R_snp_info <- read_pvar(ld_pvarf)
ld_pgen <- prep_pgen(pgenf = ldref_file, ld_pvarf)
sidx <- match(snplist_all, R_snp_info$id)
X.g <- read_pgen(ld_pgen, variantidx = sidx)
R_snp <- Rfast::cora(X.g)
colnames(R_snp) <- snplist_all
rownames(R_snp) <- snplist_all
rm(snplist_all, X.g)
####################
#prepare data object
data <- list(sum_stat=list(),
ld_mat=list())
for (j in 1:length(clumps)){
snplist <- clumps[[j]]
R_snp_current <- R_snp[snplist,snplist,drop=F]
dimnames(R_snp_current) <- NULL
R_snp_info_current <- R_snp_info[match(snplist, R_snp_info$id),c("id", "ref", "alt")]
sumstats_current <- data.frame(id=snplist,
ref=R_snp_info_current$ref,
eff=R_snp_info_current$alt,
beta_hat_eqtl=eqtl_current$slope[match(snplist, eqtl_current$SNP)],
beta_hat_gwas=gwas$Estimate[match(snplist, gwas$id)],
se_eqtl=eqtl_current$slope_se[match(snplist, eqtl_current$SNP)],
se_gwas=gwas$Std.Error[match(snplist, gwas$id)])
sumstats_current$abs_z <- abs(sumstats_current$beta_hat_eqtl/sumstats_current$se_eqtl)
data$sum_stat[[j]] <- sumstats_current
data$ld_mat[[j]] <- R_snp_current
}
par(mfrow=c(1,1))
#collapse high correlation SNP keeping most extreme z score (most significant)
data <- collapseHighCorSNPs(data$sum_stat,
data$ld_mat,
score="abs_z",
plot=F)pre: 674,226,968,12,296,268post: 37,27,74,09,18,56data <- flipAllelesAndGather(data$sum_stat,
data$ld_mat,
snp_id="id",
ref="ref",
a="eqtl",
sep="_",
b="gwas",
eff="eff",
beta="beta_hat",
se="se",
a2_plink="ref_eqtl",
alleles_same=T)
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
#colocalization
coloc_fit <- list()
nclust <- length(data$beta_hat_a)
options(mc.cores=4)
for (j in 1:nclust) {
if (length(data$beta_hat_a[[j]])>1){
coloc_fit[[j]] <- with(data,
fitBetaColoc(
beta_hat_a = beta_hat_a[[j]],
beta_hat_b = beta_hat_b[[j]],
se_a = se_a[[j]],
se_b = se_b[[j]],
Sigma_a = Sigma[[j]],
Sigma_b = Sigma[[j]]
))
} else {
coloc_fit[[j]] <- list(beta_hat_a=data$beta_hat_a[[j]],
beta_hat_b=data$beta_hat_b[[j]])
}
}Warning: There were 643 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 1369 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 1075 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 293 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 2 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.3, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 933 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2576 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 4 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.18, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 428 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: Examine the pairs() plot to diagnose sampling problemsWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 696 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 2 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.07, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essWarning: There were 452 divergent transitions after warmup. See
https://mc-stan.org/misc/warnings.html#divergent-transitions-after-warmup
to find out why this is a problem and how to eliminate them.Warning: There were 3328 transitions after warmup that exceeded the maximum treedepth. Increase max_treedepth above 10. See
https://mc-stan.org/misc/warnings.html#maximum-treedepth-exceededWarning: There were 4 chains where the estimated Bayesian Fraction of Missing Information was low. See
https://mc-stan.org/misc/warnings.html#bfmi-lowWarning: Examine the pairs() plot to diagnose sampling problemsWarning: The largest R-hat is 1.11, indicating chains have not mixed.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#r-hatWarning: Bulk Effective Samples Size (ESS) is too low, indicating posterior means and medians may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#bulk-essWarning: Tail Effective Samples Size (ESS) is too low, indicating posterior variances and tail quantiles may be unreliable.
Running the chains for more iterations may help. See
https://mc-stan.org/misc/warnings.html#tail-essres <- list(beta_hat_a = lapply(coloc_fit, `[[`, "beta_hat_a"),
beta_hat_b = lapply(coloc_fit, `[[`, "beta_hat_b"),
sd_a = data$se_a,
sd_b = data$se_b,
alleles = data$alleles)
res <- extractForSlope(res)
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
res$beta_hat_a
[1] 0.14875346 0.33998516 0.22431695 0.09251984 0.29819375 0.06850408
$beta_hat_b
[1] -0.021096848 -0.040317704 -0.024124520 -0.031645010 -0.092383982
[6] -0.005685927
$sd_a
[1] 0.0527500 0.0530704 0.1075950 0.0742875 0.0552440 0.0523450
$sd_b
[1] 0.011252839 0.011254246 0.019115350 0.012252449 0.009954775 0.010028221
$alleles
id ref eff
1 rs1860471 C T
38 rs313830 C T
126 rs2007731 G A
147 rs34430824 C T
148 rs2016325 C T
190 rs6977334 G A#sort clusters by significance
res$abs_z <- abs(res$beta_hat_a/res$sd_a)
res_sig_order <- order(-res$abs_z)
res$beta_hat_a <- res$beta_hat_a[res_sig_order]
res$beta_hat_b <- res$beta_hat_b[res_sig_order]
res$sd_a <- res$sd_a[res_sig_order]
res$sd_b <- res$sd_b[res_sig_order]
res$alleles <- res$alleles[res_sig_order,,drop=F]
res$abs_z <- NULL
####################
res_bkup <- res
####################
#trim clusters if (absolute) correlation greater than 0.05
if (length(res$alleles$id)>1){
trim_index <- trimClusters(r2=abs(R_snp[res$alleles$id,res$alleles$id,drop=F]),
r2_threshold=0.05)
if (length(trim_index)>0){
res$beta_hat_a <- res$beta_hat_a[-trim_index]
res$beta_hat_b <- res$beta_hat_b[-trim_index]
res$sd_a <- res$sd_a[-trim_index]
res$sd_b <- res$sd_b[-trim_index]
res$alleles <- res$alleles[-trim_index,,drop=F]
}
}
####################
#visual input data
par(mfrow=c(3,1))
logging::loginfo(gene)2023-06-12 15:44:06 INFO::ENSG00000237310par(mar=c(2.1, 4.1, 2.1, 2.1))
plot(df_locus$pos, abs(df_locus$z_gwas), xlab="", ylab="GWAS |Z|")
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$z_eqtl), xlab="", ylab="eQTL SS |Z|",
col=colors[df_locus$clump+1])
abline(v=df_locus$pos[df_locus$causal], col="red")
plot(df_locus$pos, abs(df_locus$weight), col="purple", xlab="", ylab="eQTL Lasso |wgt|")
abline(v=df_locus$pos[df_locus$causal], col="red")
breakpoints <- ld_R_info[ld_R_info$index!=c(ld_R_info$index[-1],max(ld_R_info$index)),]
breakpoints <- breakpoints[breakpoints$chrom==chr,]
breakpoints <- breakpoints[breakpoints$pos>min(df_locus$pos) & breakpoints$pos<max(df_locus$pos),]
abline(v=breakpoints$pos)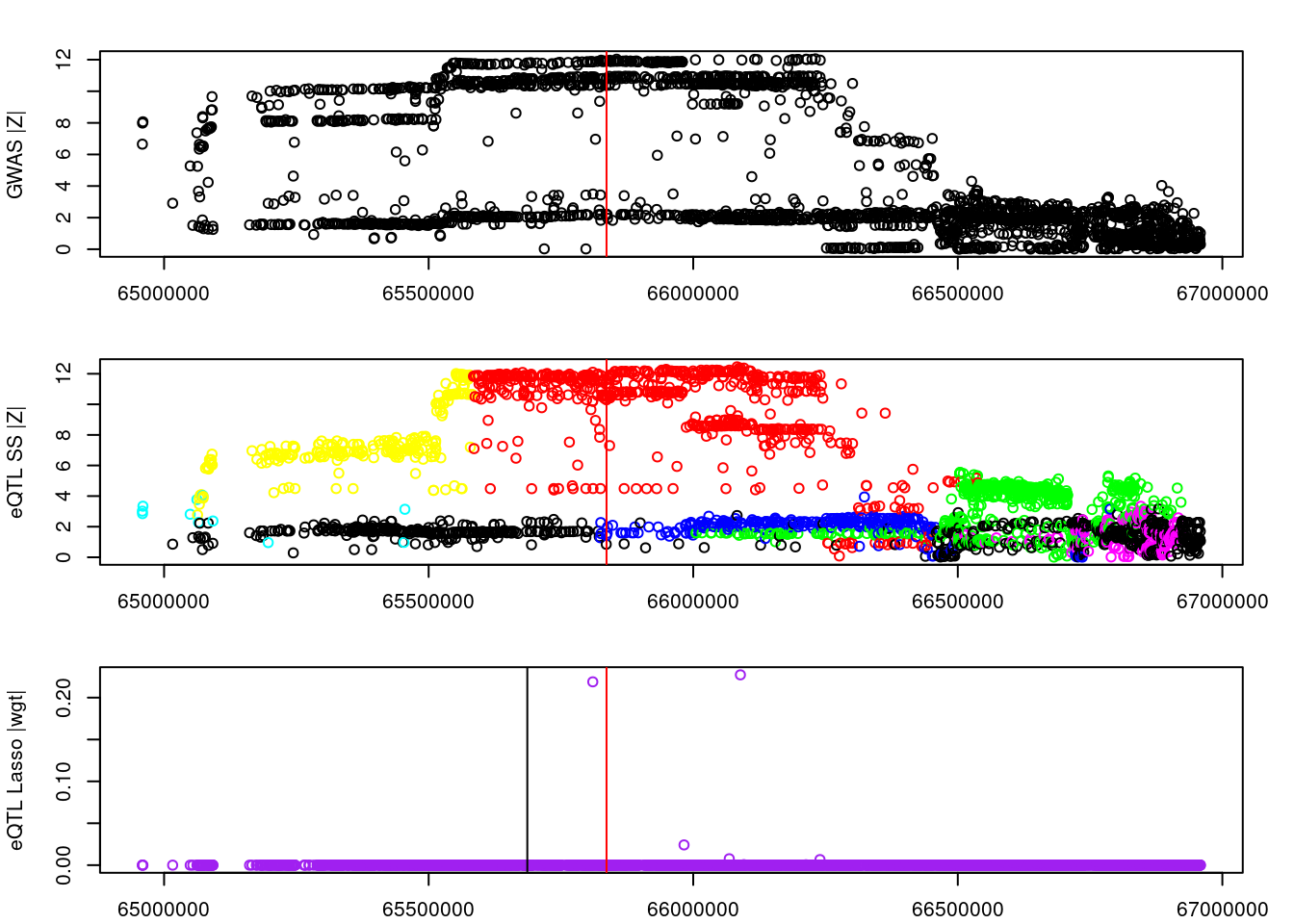
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
df_locus$trim <- F
df_locus$trim[match(res_bkup$alleles$id[trim_index], df_locus$id)] <- T
df_locus$color <- colors[df_locus$clump+1]
df_locus[match(res_bkup$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump
5526878 rs313830 12.010801 7 65551931 -10.4780020 FALSE 0 2
5528329 rs2016325 3.941677 7 66323500 -7.7619339 FALSE 0 5
5527837 rs1860471 12.451166 7 66082946 -10.4516260 FALSE 0 1
5530252 rs2007731 -4.550797 7 66825873 3.1294781 FALSE 0 3
5530376 rs6977334 -3.016926 7 66848202 0.8392996 FALSE 0 6
5526686 rs34430824 3.135588 7 65455084 -5.5923938 FALSE 0 4
trim color
5526878 FALSE #FFFF00
5528329 TRUE #0000FF
5527837 TRUE #FF0000
5530252 TRUE #00FF00
5530376 TRUE #FF00FF
5526686 TRUE #00FFFFpar(mfrow=c(1,1))
scales::show_col(colors[c(1, res_sig_order+1)])
| Version | Author | Date |
|---|---|---|
| d17186e | wesleycrouse | 2023-06-08 |
####################
if (length(res$beta_hat_a)==1){
#parametric sample from MRLocus code
m <- 1e+05
post_samples <- list(alpha=rnorm(m, res$beta_hat_b, res$sd_b)/rnorm(m, res$beta_hat_a, res$sd_a))
} else {
#fit MRLocus slope model
res <- fitSlope(res, iter=10000)
post_samples <- rstan::extract(res$stanfit)
}
par(mfrow=c(1,1))
#display results
print(res$stanfit, pars=c("alpha","sigma"), probs=c(.1,.9), digits=3)NULLif (!is.null(res$stanfit)){
plotMrlocus(res, main="MRLocus gene-to-trait effect estimate")
} else {
logging::loginfo("<2 clumps for analysis")
}2023-06-12 15:44:06 INFO::<2 clumps for analysis####################
#investigate LD between confounding variants and eSNPs for clumps
#confounding causal SNPs
df_locus[df_locus$causal,] id z_eqtl chrom pos z_gwas causal weight clump trim
5527360 rs778685 -10.57407 7 65836176 11.90402 TRUE 0 1 FALSE
color
5527360 #FF0000#eSNPs in the analysis
df_locus[match(res$alleles$id, df_locus$id),] id z_eqtl chrom pos z_gwas causal weight clump trim
5526878 rs313830 12.0108 7 65551931 -10.478 FALSE 0 2 FALSE
color
5526878 #FFFF00#LD between confounding SNPs and eSNPs
snplist_all <- c(df_locus$id[df_locus$causal], res$alleles$id)
ld_R_idx <- unique(ld_R_info$index[match(snplist_all, ld_R_info$id)])
R_snp <- lapply(ld_R_files[ld_R_idx], readRDS)
R_snp_info <- lapply(ld_R_info_files[ld_R_idx], fread)
R_snp_info <- lapply(R_snp_info, as.data.frame)
R_snp_index <- lapply(R_snp_info, function(x){x$id %in% snplist_all})
for (j in 1:length(R_snp)){
R_snp[[j]] <- R_snp[[j]][R_snp_index[[j]],R_snp_index[[j]]]
R_snp_info[[j]] <- R_snp_info[[j]][R_snp_index[[j]],,drop=F]
}
R_snp_info <- as.data.frame(do.call(rbind, R_snp_info))
R_snp <- as.matrix(Matrix::bdiag(R_snp))
colnames(R_snp) <- R_snp_info$id
rownames(R_snp) <- R_snp_info$id
#number of LD regions
length(ld_R_idx)[1] 2#LD between variants
R_snp[snplist_all, snplist_all] rs778685 rs313830
rs778685 1 0
rs313830 0 1
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /software/R-4.1.0-no-openblas-el7-x86_64/lib64/R/lib/libRblas.so
LAPACK: /software/R-4.1.0-no-openblas-el7-x86_64/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ctwas_0.1.34 mrlocus_0.0.25 data.table_1.14.6 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.9 lattice_0.20-45 prettyunits_1.1.1
[4] getPass_0.2-2 ps_1.7.2 foreach_1.5.2
[7] assertthat_0.2.1 rprojroot_2.0.3 digest_0.6.31
[10] utf8_1.2.2 R6_2.5.1 RcppZiggurat_0.1.6
[13] stats4_4.1.0 evaluate_0.18 highr_0.9
[16] httr_1.4.4 ggplot2_3.4.0 pillar_1.8.1
[19] rlang_1.0.6 rstudioapi_0.14 whisker_0.4.1
[22] callr_3.7.3 jquerylib_0.1.4 Matrix_1.5-3
[25] rmarkdown_2.18 stringr_1.5.0 loo_2.5.1
[28] munsell_0.5.0 compiler_4.1.0 httpuv_1.6.6
[31] xfun_0.35 rstan_2.21.7 pkgconfig_2.0.3
[34] pkgbuild_1.4.0 htmltools_0.5.4 tidyselect_1.2.0
[37] tibble_3.1.8 gridExtra_2.3 logging_0.10-108
[40] codetools_0.2-18 matrixStats_0.63.0 fansi_1.0.3
[43] withr_2.5.0 crayon_1.5.2 dplyr_1.0.10
[46] later_1.3.0 MASS_7.3-58.1 grid_4.1.0
[49] jsonlite_1.8.4 gtable_0.3.1 lifecycle_1.0.3
[52] DBI_1.1.3 git2r_0.30.1 magrittr_2.0.3
[55] StanHeaders_2.21.0-7 scales_1.2.1 Rfast_2.0.6
[58] RcppParallel_5.1.5 cli_3.4.1 stringi_1.7.8
[61] cachem_1.0.6 farver_2.1.1 fs_1.5.2
[64] promises_1.2.0.1 pgenlibr_0.3.2 bslib_0.4.1
[67] vctrs_0.5.1 generics_0.1.3 iterators_1.0.14
[70] tools_4.1.0 glue_1.6.2 processx_3.8.0
[73] parallel_4.1.0 fastmap_1.1.0 yaml_2.3.6
[76] inline_0.3.19 colorspace_2.0-3 knitr_1.41
[79] sass_0.4.4